286 / PRINCIPIOS GENERALES DE GENÉTICA MÉDICA
El desarrollo humano depende de factores genéticos y ambientales. La carga genética de una persona (genoma) se determina en el momento de la concepción. La información genética se localiza en el ADN de los cromosomas y de las mitocondrias. Probablemente, la mayoría de las enfermedades tienen alguna implicación genética de importancia variable. Los factores medioambientales pueden alterar la información genética mediante mutaciones u otras alteraciones estructurales y afectar a los trastornos genéticos clásicos (p. ej., tratamiento dietético en la fenilcetonuria, fármacos en la hipercolesterolemia).
La capacidad del ADN para replicarse constituye la base de la transmisión hereditaria. El ADN proporciona también el código genético, el cual determina el metabolismo y el desarrollo celular mediante el control de la síntesis de ARN. La secuencia de los elementos (nucleótidos) que forman el ADN y el ARN determina la composición proteínica y por tanto su función.
Los genes (entre 60.000 y 100.000 en el hombre) se localizan en los cromosomas (estructuras con forma de bastón presentes en el núcleo celular) y en las mitocondrias (múltiples estructuras circulares situadas en el citoplasma celular). En el hombre, las células somáticas (no embrionarias) poseen normalmente 46 cromosomas, distribuidos en 23 parejas. Cada par está formado por un cromosoma de origen materno y otro paterno. El sexo de la persona está determinado por un par, los cromosomas sexuales. La mujer tiene dos cromosomas X en cada núcleo de las células somáticas, mientras que en el hombre existe un cromosoma X y un cromosoma Y (es decir, cromosomas heterólogos). El cromosoma X es portador de genes responsables de muchos rasgos hereditarios, mientras que el cromosoma Y, más pequeño y de forma distinta, contiene los genes que inician la determinación sexual masculina. Las 22 parejas de cromosomas restantes, los autosomas, suelen ser homólogos (es decir, de idéntico tamaño, forma, posición y número de genes). Las células germinales (óvulo y espermatozoide) realizan la meiosis, la cual reduce el número de cromosomas a 23 (la mitad que las células somáticas [46]), de modo que cuando un óvulo es fertilizado por un espermatozoide se restituye el número normal de cromosomas. En la meiosis la información genética que se hereda del padre y de la madre se recombina por entrecruzamiento o intercambio entre los cromosomas homólogos.
Los genes, la unidad básica de la herencia, están dispuestos de forma lineal en el interior del ADN a lo largo de los cromosomas; cada gen tiene una localización o una posición específica (locus) en los cromosomas. El número y la disposición de un locus en el cromosoma homólogo suelen ser idénticos. No obstante, la estructura de un gen específico puede tener variaciones mínimas (polimorfismos) sin ser causa de enfermedad. La secuencia específica de nucleótidos de los genes que ocupan dos loci homólogos a lo largo de los dos cromosomas de un par se denominan alelos. Los dos alelos (es decir, uno heredado de la madre y otro del padre) pueden presentar ligeras diferencias en la secuencia de nucleótidos o ser ésta la misma. Una persona con un par de alelos idénticos de un gen determinado es un homocigoto, una persona con un par de alelos distintos es un heterocigoto.
Si se manifiesta un carácter o un trastorno cuando sólo un alelo es anómalo, se dice que este trastorno es dominante. Éste es recesivo si se manifiesta sólo cuando los dos alelos de los loci de ambos cromosomas son anómalos. Un número pequeño de genes se localizan en el ADN mitocondrial (v. más adelante). En el citoplasma existen muchas copias de ADN mitocondrial que pueden tener una estructura similar (homoplasmia) o distinta (heteroplasmia).
Existen tres tipos de trastornos genéticos: en primer lugar las mutaciones mendelianas, o alteraciones genéticas simples, que son heredadas en patrones identificables; en segundo lugar las alteraciones multifactoriales que afectan a más de un gen e interactúan con los factores medioambientales de manera no siempre reconocible, aunque se han descrito basándose en la observación (empíricamente), y en tercer lugar las anomalías cromosómicas que incluyen defectos estructurales y desviaciones del número normal (v. cap. 247). Recientemente se han descrito patrones hereditarios no habituales y mitocondriales.
Los seres humanos varían fenotípica y genotípicamente. La heterogeneidad resulta de los distintos alelos y de las mutaciones de los genes múltiples, los cuales participan en casi todas las rutas bioquímicas. Las mutaciones que se localizan en zonas diferentes de un gen pueden originar trastornos distintos. Por ejemplo, las mutaciones en distintas zonas a lo largo de un gen para un tipo de colágeno pueden producir estatura alta, artritis y sordera o enanismo. Se han identificado más de 60 causas específicas de sordera congénita; algunas son genéticas (con implicación de los genes nucleares y mitocondriales), mientras que otras son consecuencia del virus de la rubeola o de otros agentes medioambientales. Está aumentando la preocupación respecto a los efectos teratógenos de algunas drogas durante el embarazo. Por ejemplo, una mujer que consuma alcohol durante el embarazo presenta un riesgo elevado de tener descendencia con retraso mental y alteraciones del comportamiento, retraso del crecimiento intrauterino y malformaciones congénitas (v. Síndrome alcohólico fetal en Problemas metabólicos en el recién nacido en el cap. 260).
Así, trastornos fenotípicamente similares pueden estar causados por mutaciones ocurridas en genes distintos, factores no genéticos o ambos. Para establecer el origen del problema y determinar mejor el riesgo de la futura descendencia, el médico debe obtener una historia familiar completa e indagar respecto a los posibles factores medioambientales, teniendo presente la heterogeneidad. No siempre es posible identificar el agente causante.
Las potentes técnicas de genética molecular han hecho posible el estudio de la estructura del ADN y la detección de cambios durante el desarrollo y en distintos tejidos. La estructura de un gen es compleja e incluye elementos de control (p. ej., promotores, amplificadores), elementos expresados (exones), elementos mediadores que no se expresan (intrones) y señal de finalización. La configuración del ADN de un gen en un tejido que no expresa el gen es probablemente diferente (p. ej., metilado, condensado) del de un gen que se expresa de forma activa.
El proyecto genoma humano es un gran proyecto de colaboración internacional que se inició en 1991. Su objetivo hasta el 2005 es trazar un mapa de las localizaciones específicas de genes determinados en los cromosomas y determinar su secuencia exacta de nucleótidos (el genoma humano). Este trazado se puede efectuar mediante estudios familiares, utilizando la localización conocida de marcadores del ADN, e implica el aislamiento de pequeños intervalos de ADN y la secuenciación de genes y el otro ADN en esta zona.
Detección selectiva (screaning) genética
La detección selectiva (screaning) genética se puede emplear en poblaciones de riesgo para una alteración genética particular. El diagnóstico genético sólo es apropiado cuando se conoce la historia natural de la enfermedad, las pruebas diagnósticas son válidas y fiables, los índices de sensibilidad, de especificidad, de falsos negativos y de falsos positivos son aceptables y existe un tratamiento eficaz. El beneficio obtenido de un programa de diagnóstico selectivo debe ser suficiente como para justificar su coste.
Detección selectiva heterocigota. Puede estar indicado el diagnóstico selectivo en poblaciones susceptibles (p. ej., enfermedad de Tay-Sachs en judíos askenazí, anemia de células falciformes en gente de raza negra, talasemia en varios grupos étnicos) debido a la alta frecuencia de heterocigotos. La detección selectiva heterocigótica puede determinar si una persona es portadora de un trastorno específico. Si su pareja es también heterocigota, existe el riesgo de que su descendencia herede esta alteración. Este diagnóstico permite que la pareja adopte decisiones con la información previa adquirida respecto a su descendencia.
Detección selectiva genética presintomática. La detección selectiva genética presintomática puede ser apropiada en personas con una historia familiar de un trastorno familiar dominante (p. ej., enfermedad de Huntington, cáncer de mama). La identificación de un portador de un trastorno genético puede permitir que el paciente tome decisiones con la información previa adquirida (p. ej. monitorización en el caso de cáncer de mama, alternativas respecto a la reproducción en el caso de enfermedad de Huntington o de enfermedad renal poliquística del adulto).
Diagnóstico prenatal. La amniocentesis, las muestras de vellosidades coriónicas, de sangre del cordón umbilical, de sangre materna, la valoración selectiva del suero materno y la visualización fetal con ecografía o radiología son útiles en el diagnóstico prenatal (v. cap. 247). Las razones habituales de diagnóstico prenatal incluyen la edad materna superior a 35 años, la existencia de antecedentes familiares de un trastorno que es diagnosticable mediante técnicas prenatales, los resultados anómalos en el suero materno y ciertas complicaciones del embarazo.
Detección selectiva del recién nacido. La detección de la fenilcetonuria, la galactosemia y el hipotiroidismo en el recién nacido permite iniciar la profilaxis (p. ej., la administración de dietas especiales o el tratamiento sustitutivo) lo suficientemente pronto como para prevenir las complicaciones graves.
Construcción del árbol genealógico de una familia
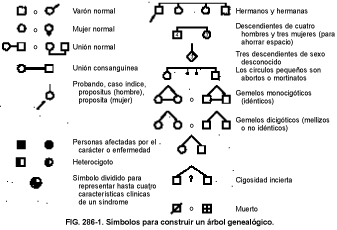
La historia familiar suele ser la clave para determinar el riesgo genético. Ésta se registra mejor en la genealogía familiar (árbol familiar), que emplea símbolos convencionales (v. fig. 286-1). El árbol genealógico proporciona una visión rápida de los problemas o las enfermedades que existen en la familia y facilita el análisis de los patrones de herencia, incluyendo la línea y el grado de afectación y las variaciones entre personas y generaciones. Algunas enfermedades familiares con fenotipos idénticos tienen varios patrones de herencia. Por ejemplo, el paladar hendido puede presentar un patrón de herencia autosómico dominante, autosómico recesivo o ligado al cromosoma X, o puede ser multifactorial (es decir, familiar sin un patrón de transmisión previsible).
Las generaciones se designan con números romanos en el árbol genealógico familiar, con las más antiguas en la parte superior y las más recientes en la base (v. figs. 286-2 a 286-5). Las personas se numeran de izquierda a derecha con números árabes dentro de cada generación. Los hermanos o hermanas se suelen presentar según la edad, situándose el mayor a la izquierda. Así, cada miembro del árbol puede ser identificado por dos números (p. ej., II, 4). Al cónyuge se le asigna también un número de identificación (p. ej., II, 6 en fig. 286-2).
El estudio de un carácter o una enfermedad comienza por la persona afectada (el probando, propositus [hombre], proposita [mujer], o caso índice). Cuando se recoge la historia familiar, el médico debe realizar el árbol conforme se van describiendo los parientes. La investigación comienza con los hermanos y hermanas del probando y luego sigue con los padres, parientes de los padres, incluyendo hermanos, hermanas, sobrinos y sobrinas, abuelos, y de ahí en adelante. El número de pacientes incluidos en el árbol genealógico viene determinado por el patrón hereditario del trastorno y por la extensión de la memoria o el conocimiento del informador. Por lo general, se deben incluir al menos 3 generaciones. También se deben registrar enfermedades, hospitalizaciones, causas de muerte, mortinatos, abortos, anomalías congénitas y cualquier otra información que se considere útil.
Consejo genético
El consejo genético consiste en la obtención de una historia familiar completa y en la evaluación de las preocupaciones y las cuestiones familiares mediante la combinación de la información apropiada recogida de la literatura y de los especialistas en genética. También suele ser importante consultar con un experto sobre cada cuestión específica. La información adquirida debe incluir el diagnóstico y los métodos diagnósticos, englobando la identificación de portadores, la historia natural de la enfermedad y sus complicaciones, el riesgo de recidiva para los pacientes y los diferentes miembros de la familia, los tratamientos posibles y las opciones de reproducción. La comunicación de los riesgos genéticos y de las opciones es un proceso delicado que suele precisar visitas de seguimiento y comunicación escrita.
Muchas familias crean grupos para el estudio de enfermedades genéticas específicas. La mayoría mantienen reuniones periódicas y publican información de utilidad.
En las facultades de medicina de Estados Unidos y Canadá existen centros de consejo genético donde se puede enviar a los pacientes y a sus familiares para realizar el diagnóstico y proporcionar consejo, tratamiento e información. Es fundamental una relación estrecha entre el médico de familia y el centro de genética por el interés de la familia y para el seguimiento de la enfermedad.
HERENCIA DE LOS DEFECTOS GENÉTICOS SIMPLES
Los trastornos genéticos determinados por un gen simple son los más fáciles de analizar y se han estudiado con mayor profundidad. Se han descrito numerosos defectos específicos. Los defectos genéticos simples pueden ser autosómicos o ligados al cromosoma X, bien dominantes o recesivos.
AUTOSÓMICA DOMINANTE
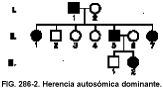
Una persona sólo necesita tener un alelo anormal de un gen para presentar un defecto de tipo autosómico dominante. En la figura 286-2 se representa el árbol genealógico característico de un rasgo autosómico dominante, en el que se produce una transmisión vertical. En general, se pueden aplicar las siguientes normas:
• Una persona afectada tiene antecesor afectado.
• Una persona afectada y una persona no afectada tienen, como promedio, un número igual de descendientes afectados y no afectados.
• Los hijos no afectados de un padre o una madre afectada tienen hijos y nietos no afectados.
• Los varones y las mujeres tienen la misma probabilidad de ser afectados.
• El riesgo de presentación entre los hijos de una persona afectada es del 50%.
El árbol genealógico se basa en el fenotipo (características observables). Con los estudios moleculares también se puede determinar y registrar el genotipo.
Expresividad y penetrancia. Los efectos de un gen pueden estar influenciados por el ambiente y por otros genes que pueden alterar la expresión fenotípica (o expresividad). Por eso, incluso dentro de una misma familia, las personas con la misma alteración de un gen (alelo) pueden manifestar fenotipos muy variables. Por ejemplo, en el síndrome de Waardenburg tipo I, una mutación en el gen PAX3 que produce de manera característica un copete (mechón) blanco, ojos separados y heterocromía del iris, pero en el que <20% de los casos tienen una pérdida auditiva significativa. Algunos miembros de la familia con el alelo anormal no presentan ninguna de estas características excepto el mechón blanco, pero sus hijos presentan sordera profunda congénita debida a la misma mutación. En casos raros, la expresividad es tan leve que no se puede detectar ninguna anomalía clínica, aunque el portador del alelo anormal no afectado se lo puede pasar a su descendencia, que puede desarrollar el cuadro clínico completo. En este caso, en el árbol genealógico parece existir una generación respetada. Este fenómeno se conoce como falta de penetrancia. Sin embargo, algunos casos de falta de penetrancia aparente se deben a la incapacidad del médico examinador para reconocer las manifestaciones menores del trastorno. Los casos con una expresividad mínima se suelen denominar como forma frustrada de la enfermedad.
Pleiotropía. Un defecto genético aislado puede producir anomalías múltiples en diferentes sistemas orgánicos. Por ejemplo, en la osteogénesis imperfecta (una anomalía del tejido conjuntivo en la que muchos pacientes presentan anomalías reconocibles en los genes del colágeno [v. también Anomalías musculoesqueléticas en el cap. 261) pueden existir huesos frágiles, sordera, color azulado de la esclerótica, dientes displásicos, hiperlaxitud articular y anomalías de las válvulas cardíacas. Debido a que todas estas anomalías afectan al colágeno de diferentes tejidos y dado que los tipos específicos de colágeno tienen una distribución previsible, el resultado es la afectación de diferentes órganos y sistemas.
Herencia limitada por el sexo. Un carácter que aparece sólo en uno de los dos sexos se denomina limitado por el sexo. Esto es diferente de la herencia ligada al cromosoma X, que se refiere a los caracteres que son transmitidos en el cromosoma X. Las hormonas sexuales y otras diferencias fisiológicas entre hombres y mujeres pueden alterar la expresividad de un gen. Por ejemplo, la alopecia prematura es un rasgo autosómico dominante pero, presumiblemente como resultado de las hormonas sexuales femeninas, el trastorno se expresa en raras ocasiones en la mujer y sólo después de la menopausia. Por estas razones, la herencia limitada por el sexo es un caso especial de expresividad y penetrancia limitadas.
Mutaciones. Son cambios en la información genética que surgen de manera espontánea. La herencia autosómica dominante comienza con una mutación reciente, en la que la información genética (ADN) heredada de los padres ha sufrido un cambio. Las tasas de mutación son variables (1/6.000 a 1/50.000 personas) entre las enfermedades autosómicas recesivas reconocidas.
Por ejemplo, alrededor del 80% de las personas con enanismo acondroplásico (un rasgo autosómico dominante) no presentan historia familiar y constituyen mutaciones espontáneas. Casi todas las personas afectadas pueden transmitir a sus descendientes el gen con la mutación reciente. En la acondroplasia, las mutaciones ocurren en una región muy específica del gen. En muchos otros trastornos, cada nueva mutación parece ocurrir en una región diferente del gen.
En general, los pacientes no afectados no presentan un riesgo elevado de tener descendientes adicionales afectados, pero algunos padres con aspecto normal han tenido dos e incluso tres descendientes con enanismo acondroplásico típico. La explicación es una mutación en la línea germinal, es decir, un evento temprano en la vida embrionaria de un padre/madre de aspecto normal cuando sólo existen unos pocos precursores de las células germinales. La célula que posee la nueva mutación puede contribuir con numerosas células a la gónada en desarrollo. En estos casos, la probabilidad de tener otro hijo afectado puede ser tan alta como el 50%. La existencia de mutaciones en la línea germinal se ha confirmado mediante estudios moleculares del gen mutado en progenitores y descendientes. En el caso del padre con un fenotipo normal portador de una mutación en la línea germinal, los estudios moleculares han confirmado que numerosos espermatozoides pueden ser portadores de la mutación.
AUTOSÓMICA RECESIVA
El árbol genealógico típico se muestra en la figura 286-3. Una persona debe tener dos copias de un alelo anormal para desarrollar una enfermedad autosómica recesiva. Ciertas poblaciones deben presentar una tasa elevada de heterocigotos o portadores debido a un efecto fundador (es decir, el grupo comenzó con pocos miembros, uno de ellos era el portador) o porque los ascendientes pueden haber recibido un portador con una ventaja selectiva (p. ej., el patrón heterocigoto para la anemia de células falciformes protege frente a la malaria).
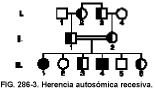
En general, se pueden aplicar las siguientes reglas de herencia:
• Si los progenitores normales tienen un descendiente afectado, ambos padres son heterocigotos, y, como promedio, el 25% de los hijos estarán afectados, el 50% serán heterocigotos y el otro 25% serán normales.
• Todos los hijos de una persona afectada y de una persona con genotipo normal serán heterocigotos con fenotipo normal.
• En promedio, el 50% de los hijos de una persona afectada y de un heterocigoto estarán afectados y la otra mitad serán heterocigotos.
• Todos los hijos de dos personas afectadas también lo estarán.
• Los hombres y las mujeres tienen las mismas probabilidades de verse afectados.
• Los heterocigotos presentan un fenotipo normal pero son portadores del carácter. Si la enfermedad está producida por un defecto en una proteína específica (p. ej., una enzima), el portador suele presentar una cantidad reducida de dicha proteína. Si la mutación es conocida, las técnicas moleculares genéticas pueden identificar a las personas heterocigotas con fenotipo normal.
La consanguinidad (es decir, la unión entre miembros de la misma familia) puede ser importante en las enfermedades autosómicas recesivas. Las personas con parentesco tienen más probabilidades de compartir el mismo alelo mutante. Se calcula que cada ser humano es heterocigoto (es decir, portador) de seis a ocho alelos que podrían, en estado homocigótico, provocar a una enfermedad. Una historia familiar detallada puede descubrir la presencia de consanguinidad desconocida u ocultada. Las uniones progenitor-descendiente o entre hermanos (denominadas incesto) presentan un riesgo elevado de descendencia anormal, porque el 50% de sus genes son iguales.
DOMINANTE LIGADA AL CROMOSOMA X
El árbol genealógico típico se muestra en la figura 286-4. En general, se pueden aplicar las siguientes reglas:
• Los varones afectados transmiten el carácter a todas sus hijas, pero no a sus hijos (no se produce transmisión varón-varón).
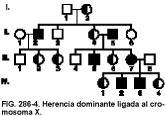
• Las mujeres heterocigóticas afectadas transmiten el rasgo a la mitad de sus descendientes, con independencia del sexo de éstos.
• Las mujeres homocigóticas afectadas transmiten el rasgo a todos sus descendientes.
• El doble de mujeres que de hombres afectados presentarán la enfermedad a menos que ésta sea mortal en varones.
Los trastornos dominantes ligados al cromosoma X, que son muy raros, suelen afectar a los varones con más gravedad que a las mujeres. Sin embargo, las portadoras de sólo un alelo anormal están afectadas. Por ejemplo, en la incontinencia pigmentaria, el gen ligado al cromosoma X es mortal en varones y produce un patrón peculiar abigarrado de pigmentación melánica y otras anomalías en dientes, ojos y SNC en mujeres. En la diabetes insípida nefrogénica, las mujeres presentan sólo grados leves de polidipsia y poliuria.
La herencia dominante ligada al cromosoma X es difícil de diferenciar de la herencia autosómica dominante sin el uso de sondas moleculares. Son necesarios árboles genealógicos muy amplios, que prestan atención especial a los descendientes de los varones afectados, ya que la transmisión varón-varón descarta la herencia ligada al cromosoma X (los varones pasan sus cromosomas Y a sus hijos varones).
RECESIVA LIGADA AL CROMOSOMA X
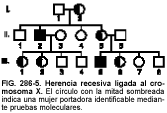
En la figura 286-5 se representa un árbol genealógico característico. En general, se pueden aplicar las siguientes normas:
• La mayoría de las personas afectadas son varones.
• Si el carácter se transmite a través de una madre heterocigótica, ésta suele presentar un fenotipo normal.
• El carácter puede representar una nueva mutación en la mujer afectada.
• Un varón afectado nunca transmite el carácter a sus hijos varones.
• Todas las hijas de un varón afectado serán portadoras.
• La mujer portadora transmite el carácter a la mitad de sus hijos varones.
• Ninguna hija de una mujer portadora presentará el carácter, pero la mitad de ellas serán portadoras (a menos que también hereden el carácter de su padre, como la ceguera para los colores).
Una mujer debe presentar el gen anormal en sus dos cromosomas X (homocigota) para que éste se exprese. Esto puede ocurrir si el padre está afectado y la madre es heterocigótica u homocigótica para el alelo mutante. Sin embargo, en varones se expresan todos los genes del cromosoma X, bien sean recesivos o dominantes. Debido a que todos los trastornos recesivos ligados al cromosoma X son raros, es muy escasa la afectación de las mujeres (la incidencia en mujeres es la raíz cuadrada de la incidencia en hombres). Además, como promedio, la mitad de los tíos maternos del probando están afectados. También la mitad de las tías maternas son portadoras, de manera que algunos de los primos hermanos maternos varones estarán afectados.
En ocasiones, las mujeres que son heterocigotas para las mutaciones ligadas al cromosoma X muestran grados variables de expresión, pero se ven afectadas en pocas ocasiones con tanta gravedad como los varones hemicigóticos. Una reordenación cromosómica estructural (p. ej., una traslocación autosoma-X, un cromosoma X perdido o borrado) puede provocar que una mujer esté afectada, incluso aunque sea heterocigótica.
CODOMINANTE
En la herencia codominante, en una persona heterocigótica se expresan ambos alelos. Debido a que por lo general ambos alelos de un locus genético producen algún producto, la codominancia, o expresión de ambos alelos, es apreciable únicamente si los fenotipos o productos de los alelos son cualitativamente diferentes, como con los antígenos de los grupos sanguíneos (AB, MN), los antígenos de los leucocitos (DR4, DR3) y las proteínas séricas que difieren en su movilidad electroforética (albúmina, haptoglobina).
HERENCIA MULTIFACTORIAL
Los parientes cercanos (hasta de tercer grado) tienden a parecerse en cierto número de características medibles o cuantitativas (altura, peso, tamaño y forma de la nariz, rasgos faciales, TA, inteligencia). Muchos caracteres se distribuyen según una curva con forma de campana, un fenómeno compatible con la determinación del carácter por varios genes diferentes. Cada gen se añade o sustrae al carácter y actúa de forma aditiva independiente de los otros genes. En los extremos de la distribución existen pocas personas y la mayoría están en el centro, porque es poco probable que una persona herede numerosos factores que actúen en la misma dirección. Los factores ambientales, cada uno añadiéndose o sustrayéndose del resultado final, también producirán una distribución normal.
Muchas anomalías congénitas y enfermedades familiares relativamente frecuentes no siguen las reglas de la herencia (mendelianas) de un gen simple. Es más probable que estas alteraciones surjan por una herencia multifactorial. Un umbral separa a la persona afectada de la no afectada. La persona afectada está predispuesta para el trastorno, representando la suma de influencias genéticas y ambientales. Por eso, el riesgo de expresión de un carácter determinado en parientes de primer grado (hermanos e hijos), que comparten el 50% de los genes de la persona afectada, es relativamente alto. El riesgo en parientes más lejanos, en quienes es probable que hereden sólo algunos genes afines, es mucho menor.
Por ejemplo, los defectos del tubo neural (DTN) (anencefalia, espina bífida, encefalocele, mielomeningocele) suelen presentar causas multifactoriales, aunque también tienen muchas causas específicas, conocidas como gen simple, cromosómicas o ambientales. En la población blanca de Estados Unidos, los DTN presentan una incidencia combinada de cerca de 1,5/1.000 nacidos vivos. Se cree que los progenitores no afectados de un lactante afectado son portadores de genes de alto riesgo y tienen una probabilidad de 1/30 (3%) de tener un segundo descendiente afectado. De manera similar, un progenitor con un DTN multifactorial tiene una probabilidad del 3 al 4% de tener un hijo afectado. En el caso raro de una pareja con dos descendientes afectados, el riesgo de tener un tercero aumenta hasta el 7 al 8%.
Los factores ambientales desempeñan un papel en la herencia multifactorial. La incidencia de DTN se acerca al 1/100 en las regiones del oeste del Reino Unido, y cuando las personas de esta área de alto riesgo emigraron a Estados Unidos, el riesgo descendió, pero se mantuvo más elevado que el de la población estadounidense. Además, se pueden prevenir entre el 50 y el 70% de los DTN mediante suplementos maternos de ácido fólico (400 mg/día) desde 1 mes antes de la concepción hasta 3 meses después de la misma. Aunque la deficiencia materna de ácido fólico tiene un papel importante, no es la única causa ambiental de DTN.
Otros ejemplos de herencia multifactorial, con riesgos similares para los hermanos y descendientes de personas afectadas, son las cardiopatías congénitas (v. cap. 261), la epilepsia idiopática (pequeño mal o gran mal) y la mayoría de los casos de labio leporino con paladar hendido o sin él. En la estenosis congénita del píloro, la relación hombre:mujer es de 5:1, lo que sugiere que el umbral es más alto para las mujeres. Por eso, la mujer precisa genes de alto riesgo más potentes que el hombre para desarrollar la enfermedad, tiene mayor número de hermanos afectados y tiene un riesgo más elevado de tener descendientes con la enfermedad.
La atención se está centrando en las enfermedades frecuentes del adulto con causas multifactoriales (p. ej., hipertensión, cardiopatía arterioesclerótica, diabetes mellitus, cáncer, artritis). Se están encontrando numerosos genes específicos. Los factores predisponentes determinados genéticamente, incluidos la historia familiar y los parámetros bioquímicos y moleculares, pueden servir para identificar a las personas de riesgo que se podrán beneficiar con mayor probabilidad de las medidas preventivas.
HERENCIA NO TRADICIONAL
Además de los patrones tradicionales de herencia descritos previamente, se han descrito otros mecanismos que implican estructuras determinadas genéticamente y pueden producir diferentes trastornos.
Mosaicismo. El mosaicismo es la presencia de dos o más líneas celulares que difieren en el genotipo o el cariotipo pero derivan de un cigoto. Es probable que ocurran mutaciones durante las divisiones celulares de cualquier organismo multicelular incluso aunque el aparato genético de la división celular suele ser muy preciso y existen numerosos mecanismos para reparar los errores provocados durante la replicación. Se calcula que cada vez que se divide una célula se producen entre cuatro y cinco cambios en el genoma. Por esta razón, cualquier organismo multicelular tendrá subclones de células con una carga genética ligeramente diferente. Estas mutaciones somáticas (mutaciones durante la división celular mitótica) pueden no conducir a ninguna enfermedad, pero sí a trastornos en los que se producen cambios salteados. Las técnicas de genética molecular han demostrado mutaciones en las células anormales implicadas cuando se las compara con los tejidos normales de soporte que las rodean. Por ejemplo, en el síndrome de McCune-Albright existen cambios displásicos salteados en el hueso, anomalías de las glándulas endocrinas, cambios pigmentarios parcheados y anomalías del corazón e hígado en algunas ocasiones. Las personas con estas anomalías en todas las células morirían, por lo que el trastorno no pasaría a la generación siguiente, pero estos pacientes sobreviven porque existe tejido normal junto al tejido patológico. En ocasiones, en un trastorno por un gen simple, un progenitor parece tener una forma leve, aunque se trata en realidad de un mosaico. Sus descendientes más afectados habrán recibido una célula germinal con el alelo mutante y por esta razón presentan las mismas anomalías en todas sus células. El mosaicismo cromosómico ocurre en algunos embriones y se puede demostrar en la placenta en muestras de vellosidades coriónicas. La mayoría de los embriones y fetos con anomalías cromosómicas se transforman en abortos de manera espontánea. No obstante, el desarrollo de células normales puede compensar ciertas anomalías cromosómicas, permitiendo que los descendientes sobrevivan.
Impronta genómica. La impronta genómica es la expresión diferencial del material genético dependiendo de si se ha heredado de la madre o del padre. La impronta genómica es específica del tejido y específica del momento del desarrollo. La expresión bialélica o biparental de los alelos puede estar presente en algunos tejidos mientras que en otros la expresión puede ser uniparental. El síndrome de Angelman y el síndrome de Prader-Willi pueden estar producidos por deleciones en el cromosoma 15. Existen grupos de genes específicos en íntima proximidad en el cromosoma 15 con expresión uniparental materna o paterna. Dependiendo de que el cromosoma que presente la deleción sea el heredado de la madre o el del padre, se producirá un síndrome diferente.
Muchas áreas de varios cromosomas tienen este tipo de origen del efecto según el progenitor. Los genes implicados parecen estar relacionados con el crecimiento y la conducta en las fases iniciales del desarrollo. Algunos de estos genes también están implicados en neoplasias benignas y malignas. Se debe tener en cuenta la posibilidad de impronta genómica en enfermedades que parecen respetar a toda una generación.
Disomía uniparental. La disomía uniparental se produce
cuando los dos cromosomas de un
par se heredan de un solo progenitor. Este fenómeno es muy raro y se cree que
implica el rescate de una trisomía, es decir, el cigoto comenzó como trisomía y
se perdió uno de los tres cromosomas, lo que conduce a una disomía uniparental
en un tercio de los casos. Se pueden ver efectos de la impronta porque está
ausente la información genética procedente del otro progenitor. Además, si el
mismo cromosoma está duplicado (isodisomía) y este cromosoma es portador de un
alelo anormal para una enfermedad autosómica recesiva, una persona con una
disomía uniparental puede presentar una enfermedad autosómica recesiva aunque
sólo uno de los progenitores sea portador. En presencia de una disomía
uniparental se debe considerar la presencia de anomalías vestigiales en los
cromosomas de algunos tejidos.
Repetición de triplete, mutaciones inestables. Un
triplete repetido es una forma rara de mutación en la que un triplete de
nucleótidos
aumenta en número en el interior de un gen (un gen normal tiene relativamente
pocos tripletes repetidos). Se ha detectado este tipo de mutación en varias
enfermedades, en especial en las que afectan al SNC. Cuando se transmite el gen
de una generación a la siguiente, o en ocasiones cuando se dividen las células
dentro del organismo, el triplete repetido puede expandirse y crecer hasta un
punto en que el gen deja de funcionar con normalidad. Los ejemplos son la
distrofia miotónica, la enfermedad de Huntington, el retraso mental
con X frágil y otros trastornos neurológicos. El número de repeticiones puede
aumentar de manera exagerada en la formación de las células germinales o en
ciertos tejidos a medida que se desarrollan el embrión y el feto. La expansión
puede ser aún mayor cuando se transmite de un progenitor (la madre en la
distrofia miotónica, el padre en la enfermedad de Huntington). Por estas razones
se puede comprobar un efecto de «progenitor de origen» y de anticipación. Este
tipo de mutación se detecta mediante estudios moleculares.
Anticipación. La anticipación se produce cuando un trastorno aparece a una edad más temprana y con mayor gravedad en cada generación sucesiva. Puede ocurrir porque un progenitor sea un mosaico y el descendiente tenga la mutación completa en todas sus células. La expansión de la repetición del triplete puede demostrar anticipación cuando aumenta el número de repeticiones con cada generación.
TRASTORNOS CROMOSÓMICOS
Los genes son transportados en los cromosomas, por esta razón, la pérdida o la presencia de cromosomas extra puede tener un efecto sustancial sobre el funcionamiento de los genes en dicha región. Las familias de genes con la misma función se pueden agrupar en un cromosoma o pueden estar dispersas en muchos cromosomas diferentes. En el capítulo 261 se exponen las anomalías cromosómicas específicas.
ANOMALÍAS DEL ADN MITOCONDRIAL
Las mitocondrias son organelas intracelulares que generan energía mediante una serie de complejos de cadena respiratoria. Contienen un cromosoma circular único que codifica 13 proteínas, varios ARN y algunas enzimas reguladoras. Sin embargo, más del 90% de las proteínas mitocondriales están codificadas por genes nucleares. Cada célula contiene varios cientos de mitocondrias en su citoplasma.
Las enfermedades mitocondriales se deben a anomalías del ADN
mitocondrial (deleciones,
duplicaciones, mutaciones). Los tejidos de alta energía, como el músculo, el
corazón y el cerebro, tienen un riesgo más elevado, pero el oído, el hígado y el
páncreas también lo tienen. Los tipos de afectación tisular se correlacionan con
cambios específicos del ADN mitocondrial, por ejemplo la oftalmoplejía externa
progresiva; su variante, el síndrome multisistémico de Kearns-Sayre
(oftalmoplejía externa progresiva crónica, bloqueo cardíaco, retinitis
pigmentaria, degeneración del SNC); el síndrome de Pearson (anemia
sideroblástica, insuficiencia pancreática y hepatopatía progresiva que comienza
en los primeros meses de vida y suele ser mortal en lactantes); la neuropatía
óptica hereditaria de Leber (una pérdida visual variable, pero a menudo
devastadora, que suele afectar a adolescentes y se debe a una mutación puntual
en el ADN mitocondrial); MERRF (Epilepsia mioclónica, fibras musculares
rojas rasgadas, demencia, ataxia, miopatía, del inglés Myoclonic Epilepsy,
Ragged Red Fibers, dementia, ataxia, myopathy), y MELAS
(encefalomiopatía mitocondrial, acidosis láctica, episodios seudosincopales, del
inglés Mitochondrial Encephalomyopathy, Lactic Acidosis,
Strokelike episodes). La patología mitocondrial está presente en
numerosos trastornos frecuentes (deleciones mitocondriales amplias en las
células de los ganglios basales de pacientes con enfermedad de Parkinson, muchos
tipos de miopatías, acumulación progresiva de deleciones en el ADN mitocondrial
con la edad).
La herencia materna caracteriza las anomalías del ADN mitocondrial, ya que todas las mitocondrias se heredan a partir del óvulo. Por este motivo, todos los descendientes de una mujer afectada tienen riesgo de heredar la anomalía, mientras que los descendientes de un varón afectado no presentan este riesgo. La variabilidad de las manifestaciones clínicas es la norma y se puede deber en parte a mezclas variables de genomas mitocondriales normales y mutantes (heteroplasmia) en el interior de las células y los tejidos.
GENÉTICA DEL CÁNCER
El cáncer engloba numerosos trastornos caracterizados por el crecimiento celular incontrolado debido a cambios en la información genética en el tejido maligno. Se han reconocido dos clases principales de genes: genes supresores de tumores, que impiden el crecimiento tumoral; y oncogenes, que favorecen el crecimiento tumoral (v. también el cap. 142). Los oncogenes se originan a partir de protooncogenes o genes involucrados en la regulación del crecimiento celular.
La mayoría de los procesos malignos implican la pérdida de múltiples mecanismos de control, generalmente por pérdida cromosómica o activación de genes que están inactivos en condiciones normales. Ciertos cánceres familiares presentan una herencia de un gen mutado o con un cambio en su material genético con pérdida del alelo normal en el tejido neoplásico. Muchos genes específicos desempeñan un papel importante en el cáncer, como el gen del retinoblastoma, varios genes del tumor de Wilms y al menos dos genes del cáncer de mama. Además, ciertos trastornos, como la neurofibromatosis y el síndrome de Von Hippel-Lindau, predisponen a determinados tipos de cáncer.
INMUNOGENÉTICA
La base genética del sistema inmunológico es muy compleja. Varias inmunodeficiencias genéticas implican a diferentes partes del mecanismo inmunológico de defensa. El complejo mayor de histocompatibilidad es importante en el autorreconocimiento. La herencia del locus mayor de histocompatibilidad es autosómica recesiva, por lo que los hermanos tienen una probabilidad del 25% de presentar el mismo locus mayor de histocompatibilidad. El locus de histocompatibilidad es importante en el trasplante de órganos y juega un papel principal en la tolerancia de los trasplantes de órganos y médula ósea. Otros componentes del sistema inmunológico son las moléculas de superficie de los hematíes, que producen reacciones inmunes en la transfusión de sangre. Los sistemas de antígenos ABO y Rh de los hematíes son especialmente importantes en las reacciones transfusionales y en la incompatibilidad maternofetal.
Un clon deriva de una célula somática en lugar de proceder de la fusión de los gametos (óvulo y espermatozoide). Recientemente se ha conseguido la clonación de mamíferos a partir de células simples. Los chimeras son individuos con células de diferente origen genético (trasplante, injerto, fusión embrionaria). Por el contrario, los gemelos monocigóticos son dos individuos que se han desarrollado tras la fertilización de un óvulo por un espermatozoide.
GENÉTICA FORENSE
Las técnicas nuevas de genética molecular son muy útiles para identificar la carga genética de una persona. Se pueden utilizar numerosos marcadores genéticos -cada uno de los cuales tiene una amplia variabilidad normal entre personas diferentes- para identificar si dos personas están relacionadas genéticamente. Estos marcadores polimórficos se usan en las técnicas de ADN para identificar si una persona desciende de otra. La mitad de los marcadores deben proceder de la madre y la otra mitad del padre. Por estas razones, el análisis de muestras de sangre y de extractos de ADN permite determinar si el padre o la madre putativos de una persona determinada son también los progenitores biológicos. Generalmente se puede determinar la paternidad con precisión, excepto en el caso de gemelos monocigóticos, que presentan marcadores de ADN idénticos.
Los mismos marcadores de ADN polimórficos se pueden emplear para identificar una muestra y determinar la persona de la que procede. Se pueden usar técnicas de biopsia, muestras patológicas, extensiones de sangre, muestras de semen, tejidos subungueales e incluso cabellos. Mediante la reacción en cadena de la polimerasa, se puede amplificar el ADN de una célula simple, consiguiendo una cantidad suficiente para determinar el origen del ADN.
TRATAMIENTO GENÉTICO
El tratamiento de las enfermedades genéticas suele ser muy similar a la de otros tipos de enfermedades. Las dietas especiales pueden evitar sustancias que son tóxicas para los pacientes, como en la fenilcetonuria y la homocistinuria. Las vitaminas y otros agentes pueden mejorar una vía metabólica y reducir así los niveles tóxicos de una sustancia. Por ejemplo, el ácido fólico reduce los niveles de homocisteína en las personas portadoras de polimorfismo para la 5,10 metileno-tetrahidrofolato reductasa. El tratamiento puede requerir la reposición de una deficiencia o el bloqueo de una vía metabólica hiperactiva. A veces se puede tratar al feto tratando a la madre (p. ej., corticoides para la hipoplasia adrenal virilizante congénita) o empleando terapia celular intraútero (trasplante de médula ósea). Un recién nacido con un trastorno genético puede ser candidato a tratamiento mediante trasplante de órgano o de médula ósea.
El tratamiento genético puede consistir en la inserción de copias normales de un gen en el interior de las células de personas con un defecto genético específico. Se han realizado ensayos de tratamiento genético somático para diferentes trastornos genéticos (p. ej., deficiencia de adenosindeaminasa). El tratamiento genético germinal puede implicar la corrección de una alteración en los genes del espermatozoide o del óvulo, por lo que se considera una manera inadecuada de tratar las enfermedades genéticas por cuestiones éticas, económicas, falta de investigación en seres humanos, ausencia de conocimiento sobre si los cambios se mantendrán en el embrión en crecimiento y la facilidad relativa del tratamiento somático cuando sea necesario. El tratamiento genético puede incluir también la desactivación de genes mediante ADN invertido.