
3 / DEFICIENCIA, DEPENDENCIA Y TOXICIDAD VITAMÍNICAS
En las sociedades tecnológicamente avanzadas, la deficiencia de vitaminas es consecuencia sobre todo de pobreza, alimentación caprichosa, uso incorrecto de fármacos (v. Interacciones de los fármacos con los nutrientes, cap. 1), alcoholismo crónico o alimentación parenteral prolongada.
La dependencia de las vitaminas es consecuencia de un defecto genético en el metabolismo de la vitamina o en la unión de la coenzima relacionada con la vitamina a su apoenzima. En algunos casos, dosis de vitaminas tan elevadas como 1.000 veces las cantidades dietéticas recomendadas (CDR) mejoran la función de la vía metabólica alterada. Se han identificado personas con dependencia vitamínica para vitamina D, tiamina, niacina, vitamina B6, biotina y vitamina B12 (v. tabla 1-2). La deficiencia de vitamina B12 y la deficiencia de ácido fólico se exponen en Anemias macrocíticas megaloblásticas, cap. 127.
El tratamiento con altas dosis de vitaminas es una causa de toxicidad vitamínica (hipervitaminosis) para las vitaminas A, D, E, C y B6, para la niacina y para el ácido fólico (folato).
Las fuentes, las dosis terapéuticas habituales y las necesidades dietéticas se enumeran en las tablas 1-2, 1-3 y 1-4.
DEFICIENCIA DE VITAMINA A
La vitamina A (retinol) es liposoluble y se encuentra principalmente en aceites de hígado de pescado, hígado, yemas de huevo, mantequilla y nata. Las verduras de hojas verdes y las hortalizas amarillas contienen ß-caroteno y otros carotenoides provitamínicos que se convierten en retinal en las células mucosas del intestino delgado. El retinal es reducido a retinil y posteriormente esterificado. La mayor parte de la vitamina A del organismo se almacena en el hígado en forma de retinilpalmitato. Se libera a la circulación en forma de retinol unido a una proteína fijadora de retinol y a la prealbúmina (transtiretina). El isómero 11-cis del retinal (aldehído de la vitamina A) se combina con la opsina para formar rodopsina, el grupo prostético de los pigmentos fotorreceptores en la retina. En las células somáticas, el retinol es convertido a ácido retinoico, que se combina con receptores que se unen al ADN y regulan la expresión de los genes que mantienen los tejidos epiteliales y guían la diferenciación de otros diversos tejidos.
Las equivalencias biológicas para dietas con diferentes proporciones de retinol y ß-caroteno son las siguientes: 1 USP equivale a 1 UI; 1 UI equivale a 0,3 mg de retinol; 1 mg de ß-caroteno equivale a 0,167 mg de retinol. Otros carotenoides provitamínicos tienen la mitad de actividad del ß-caroteno.
Los análogos vitamínicos sintéticos (retinoides) se emplean cada vez más en dermatología. Se está investigando el posible papel protector del ß-caroteno, el retinol y los retinoides contra algunos cánceres epiteliales.
Etiología
La deficiencia primaria de vitamina A suele ser causada por privación dietética prolongada. Es endémica en áreas, como el sur y el este de Asia, donde el arroz, que carece de caroteno, es la dieta básica.
La deficiencia secundaria de vitamina A puede deberse a insuficiencia de conversión de caroteno en vitamina A o a interferencia en la absorción, el almacenamiento o el transporte de vitamina A. La interferencia con la absorción o el almacenamiento es probable en la enfermedad celíaca, el esprúe, la fibrosis quística, las pancreatopatías, la derivación duodenal, la obstrucción parcial congénita del yeyuno, la obstrucción de las vías biliares, la giardiasis y la cirrosis. La deficiencia de vitamina A es común en la malnutrición proteicoenergética (marasmo o kwashiorkor), principalmente porque la dieta es deficiente, pero también porque el almacenamiento y el transporte de vitamina A son defectuosos.
Síntomas y signos
La gravedad de los efectos de la deficiencia de vitamina A es inversamente proporcional a la edad. El retraso del crecimiento es un signo habitual en los niños. La ingesta o la utilización insuficientes de vitamina A pueden causar disminución de la adaptación a la oscuridad y ceguera nocturna; xerosis de la conjuntiva y la córnea; xeroftalmía y queratomalacia; queratinización de pulmón, tracto GI y epitelios del tracto urinario; aumento de la susceptibilidad a las infecciones, y a veces la muerte. Es frecuente la hiperqueratosis folicular de la piel.
Los cambios patognomónicos se limitan al ojo (v. Queratomalacia, cap. 96). El cambio más temprano, la disfunción de bastones, se puede detectar en la adaptometría a la oscuridad, la escotometría de bastones o la electrorretinografía (estas pruebas requieren sujetos cooperadores). La disfunción de la retina va seguida por cambios en la estructura y la función de las células epiteliales. La xerosis de la conjuntiva bulbar consiste en sequedad, engrosamiento, formación de arrugas y pigmentación turbia; la córnea se vuelve xerótica, infiltrada y turbia en una etapa temprana. Rápidamente sobreviene queratomalacia con licuefacción de parte de la córnea, o de toda ella, lo que conduce a rotura, con extrusión del contenido del ojo y retracción consecutiva del globo ocular (phthisis bulbi), o a abultamiento anterior (ectasia corneal y estafiloma anterior) y a ceguera. En la deficiencia avanzada se presentan las manchas de Bitot (placas espumosas superficiales compuestas por residuos epiteliales y secreciones en la conjuntiva bulbar expuesta); se deben muy probablemente a deficiencia de vitamina A cuando se presentan en niños pequeños que tienen otras indicaciones de deficiencia de vitamina A. En la deficiencia grave de vitamina A en los niños la mortalidad puede ser del 50% o más.
Datos de laboratorio y diagnóstico
La evidencia de una depleción de vitamina A no puede obtenerse en la etapa preclínica, excepto si existe una historia de ingesta insuficiente. Los niveles plasmáticos de retinol caen tras el agotamiento de los depósitos hepáticos. El rango normal es de 20 a 80 mg/dl (0,7 a 2,8 m mol/l); 10 a 19 mg/dl (0,35 a 0,66 m mol/l) es bajo, y <10 mg/dl (<0,35 m mol/l) es deficiente. La media de la proteína fijadora de retinol (PFR) en plasma es 47 mg/ml en varones adultos y 42 mg/ml en mujeres adultas. Hasta la edad de 10 años, el rango es de 20 a 30 mg/ml. Los niveles plasmáticos de vitamina A y PFR descienden en los estados de deficiencia y en las infecciones agudas. Deben excluirse otras causas de ceguera nocturna. La infección secundaria puede complicar las alteraciones corneales. El ensayo con dosis terapéuticas de vitamina contribuye a hacer el diagnóstico.
Profilaxis
La xeroftalmía es la principal causa de ceguera en los niños pequeños en la mayoría de los países en vías de desarrollo, donde se aconsejan dosis profilácticas de 66.000 mg (200.000 UI) de palmitato de vitamina A en aceite por vía oral una vez cada 3 a 6 meses para todos los niños de 1 a 4 años de edad; la dosis se reduce a la mitad para los niños menores de un año. La dieta debe incluir hortalizas de hojas verdes y frutos amarillos, como el mango y la papaya. Pan, azúcar y glutamato monosódico se enriquecen con vitamina A. Para la deficiencia secundaria deben administrarse rutinariamente suplementos de vitamina A. A los lactantes sospechosos de alergia a la leche se les debe administrar suficiente vitamina A en la fórmula sustitutiva.
Tratamiento
Se debe corregir la causa y administrar vitamina A en dosis terapéuticas inmediatamente. Suele ser eficaz el palmitato de vitamina A en aceite por vía oral en dosis de 20.000 mg (60.000 UI) al día durante 2 días, y una vez antes del alta del hospital a los 7 a 10 días. En presencia de vómitos o malabsorción, debe administrarse vitamina A hidrosoluble i.m. (las preparaciones oleosas no se usan por vía i.m.). Posteriormente, se administra de 3.200 a 8.000 mg (10.000 a 25.000 UI)/día por vía oral, repartidos en tres dosis, en forma de aceite de hígado de bacalao, aceite de palma roja u otro concentrado. Debe evitarse la administración diaria de grandes dosis, especialmente en los lactantes, porque pueden resultar tóxicas.
En el embarazo y la lactancia, las dosis profilácticas o terapéuticas no deben exceder el doble de las CDR, para evitar posibles daños al feto.
TOXICIDAD DE VITAMINA A
La ingesta excesiva de vitamina A puede causar toxicidad aguda o crónica. La toxicidad aguda en los niños puede producirse por tomar grandes dosis (>100.000 mg o 300.000 UI); se manifiesta con aumento de la presión intracraneal y vómitos, que pueden llevar a la muerte si no se interrumpe la ingestión. Tras la interrupción, la recuperación es espontánea, sin daño residual; sólo se han descrito dos casos mortales. A las pocas horas de ingerir varios millones de unidades de vitamina A comiendo hígado de oso o de foca polar, los exploradores del Ártico experimentaron somnolencia, irritabilidad, cefalea y vómitos, con descamación posterior de la piel. Las tabletas que contienen dosis masivas de vitamina A han inducido a veces toxicidad aguda al ingerirse durante mucho tiempo.
La intoxicación crónica en niños mayores y adultos suele aparecer tras dosis >33.000 mg (100.000 UI)/día tomadas durante meses. En lactantes a los que se administran 6.000 a 20.000 mg (20.000 a 60.000 UI)/día de vitamina A hidrosoluble, la evidencia de toxicidad puede aparecer en pocas semanas. Se han descrito defectos congénitos en hijos de mujeres que recibían ácido 13-cis-retinoico (isotretinoína) para ciertas enfermedades de la piel durante la gestación (v. Fármacos durante el embarazo, cap. 249).
Dosis masivas (50.000 a 120.000 mg o 150.000 a 350.000 UI) de vitamina A o sus metabolitos se administran diariamente a personas con acné globular. Aunque el tratamiento es eficaz, pone al Paciente en riesgo de toxicidad por vitamina A.
Aunque el caroteno se metaboliza en el organismo a vitamina A con una velocidad lenta, la ingestión excesiva de caroteno no causa intoxicación por vitamina A, pero produce carotinemia (niveles sanguíneos de caroteno >250 mg/dl [>4,65 m mol/l]). Esta situación suele ser asintomática, pero puede conducir a carotenosis, en la cual la piel (pero no la esclerótica) adquiere un color amarillo intenso, especialmente en las palmas de las manos y en la planta de los pies. La carotenosis también puede presentarse en la diabetes mellitus, el mixedema y la anorexia nerviosa, posiblemente por una nueva reducción de la tasa de conversión de caroteno a vitamina A.
Síntomas, signos y diagnóstico
Los signos tempranos son cabello grueso y escaso, alopecia de las cejas, piel áspera y seca y labios agrietados. Más tarde destacan la cefalea intensa, el seudotumor cerebral y la debilidad generalizada. La hiperostosis cortical y la artralgia son frecuentes, especialmente en los niños. Puede haber hepatomegalia y esplenomegalia.
Los niveles de retinol plasmático normal en ayunas oscilan desde 20 a 80 mg/dl (0,7 a 2,8 m mol/l). En la hipervitaminosis A, los niveles plasmáticos en ayunas pueden superar los 100 mg/dl (3,49 m mol/l) y a veces hasta los 2.000 mg/ml (69,8 m mol/l). El diagnóstico diferencial puede ser difícil, porque los síntomas son variados y abigarrados, pero entre ellos suele haber cefalea y eritema.
Pronóstico y tratamiento
El pronóstico es excelente en adultos y niños. Los síntomas y los signos suelen desaparecer en 1 a 4 semanas tras suspender la ingestión. No obstante, el pronóstico para el feto de una madre que toma dosis masivas de vitamina A es reservado.
DEFICIENCIA Y DEPENDENCIA DE VITAMINA D
Esta vitamina liposoluble existe principalmente en dos formas: ergocalciferol (ergosterol activado, vitamina D2), que se encuentra en la levadura irradiada, y colecalciferol (7-deshidrocolesterol activado, vitamina D3), que se forma en la piel humana por exposición a la luz solar (radiación ultravioleta) y se encuentra principalmente en aceites de hígado de pescado y yemas de huevo. La leche se enriquece con ambas formas. La síntesis en la piel es normalmente la fuente principal. Un microgramo de vitamina D equivale a 40 UI.
La vitamina D es una prohormona con varios metabolitos activos que se comportan como hormonas. La provitamina D3 se sintetiza fotoquímicamente en la piel a partir de 7-deshidrocolesterol y se isomeriza lentamente a vitamina D3, la cual es transportada por la proteína fijadora de vitamina D. En el hígado, la vitamina D3 es convertida a 25(OH)D3, la principal forma circulante. Ésta pasa a la circulación enterohepática y es reabsorbible en el intestino. Después es hidroxilada de nuevo, sobre todo en los riñones, a la forma mucho más activa metabólicamente, 1,25(OH)2D3 (1,25-dihidroxicolecalciferol, calcitriol, vitamina D hormonal). La principal función de la vitamina D hormonal es aumentar la absorción de calcio en el intestino y promover la formación y la mineralización del hueso. Estas funciones están mediadas por un receptor de vitamina D que es un factor de transcripción, el cual a su vez es el instrumento para poner en actividad un conjunto de genes que expresan la actividad biológica de la vitamina D hormonal. La 1-hidroxilación crítica del 25(OH)D3 está fuertemente estimulada por la hormona paratiroidea (PTH) y, con independencia de ésta, por la hipofosfatemia. Las acciones de la vitamina D y sus metabolitos se resumen en la tabla 3-1.
La vitamina D se emplea para tratar la osteodistrofia renal causada por la insuficiencia renal crónica (v. cap. 222).
La enfermedad ósea metabólica resultante de la deficiencia de vitamina D se denomina raquitismo en los niños y osteomalacia en los adultos. Estas enfermedades se producen por factores patogénicos comunes, pero son distintas en su expresión clínica y patológica a causa de las diferencias entre los huesos en crecimiento y los huesos maduros.
Etiología
Para la aparición de una deficiencia de vitamina D en la clínica suele ser imprescindible una exposición insuficiente a la luz solar y una ingesta dietética baja. El raquitismo no es raro en los trópicos, debido a que los lactantes están vestidos y las mujeres y los niños están confinados en casa. El raquitismo de origen nutricional es raro en los Estados Unidos, pero no infrecuente entre los inmigrantes hindúes en Gran Bretaña, donde la falta de luz solar, la quelación del calcio por el consumo de sus dietas de cereales tradicionales y la escasa ingesta de leche son probablemente los causantes. En ocasiones la causa puede residir en una ingesta muy reducida de calcio o fósforo.
La deficiencia de vitamina D también puede ser causada por defectos en la producción de 25(OH)D3 o en la acción de la 1,25(OH)2D3
(v. tabla 3-2). La deficiencia puede presentarse en el hipoparatiroidismo (v. Hipocalcemia en Trastornos del metabolismo del calcio, cap. 12); enfermedades hereditarias, como el raquitismo hipofosfatémico familiar (resistente a la vitamina D), una enfermedad dominante ligada al cromosoma X (v. Anomalías en el transporte renal, cap. 261), y en varias otras enfermedades. Algunas enfermedades interfieren en la absorción de la vitamina D o con la formación de sus metabolitos activos. La deficiencia de metabolitos de la vitamina D desemboca en estados de resistencia a la vitamina D.

Raquitismo y osteomalacia pueden aparecer cuando el aporte de vitamina D es insuficiente, su metabolismo es anormal o los tejidos son resistentes a su acción. Es importante clasificar el raquitismo y la osteomalacia según su etiología en relación con las manifestaciones de la enfermedad y el tratamiento eficaz.
Anatomía patológica
Las alteraciones en los niños incluyen calcificación escasa del hueso en crecimiento e hipertrofia de los cartílagos epifisarios. Las células del cartílago epifisario dejan de degenerar normalmente, pero el cartílago nuevo continúa formándose, por lo que el espesor del cartílago epifisario aumenta irregularmente. La calcificación cesa entonces y se acumula material osteoide alrededor de los capilares de la diáfisis. En la deficiencia crónica puede haber reabsorción de hueso esponjoso de la diáfisis y del hueso cortical.
El tratamiento suficiente con vitamina D hace posible el depósito de calcio y fosfato por medio de la degeneración de las células cartilaginosas en el curso de 24 horas y la penetración por una red vascular en 48 horas. Deja de formarse material osteoide en la diáfisis y se reanuda la producción endocondrial normal de hueso neoformado.
Los cambios son similares en los adultos, pero no se limitan a los extremos de los huesos largos.
Síntomas y signos
La osteomalacia materna puede llevar a lesiones metafisarias y a tetania del recién nacido. Los lactantes pequeños están inquietos y duermen poco. Tienen reducida la mineralización del cráneo (craneotabes) en zonas alejadas de las suturas. En los lactantes mayores, se retrasan la sedestación y el gateo, así como el cierre de las fontanelas, y existe abombamiento del cráneo y nódulos costocondrales (rosario raquítico). En los niños de 1 a 4 años se agrandan los cartílagos epifisarios en los extremos inferiores de radio, cúbito, tibia y peroné, aparece cifoescoliosis y se retrasa la marcha. En niños mayores y adolescentes, la marcha es dolorosa y, en casos extremos, se desarrollan deformaciones, como las piernas combadas y el genu valgum.
La tetania raquítica es causada por hipocalcemia y puede acompañar a la deficiencia de vitamina D infantil y del adulto. Los datos clínicos se exponen en Hipocalcemia en Alteraciones del metabolismo del calcio, cap. 12.
Las alteraciones óseas, visibles en radiografías, preceden a los signos clínicos, se hacen evidentes en el tercer o cuarto mes de vida, e incluso en el momento del nacimiento si la madre es deficiente en vitamina D. Las alteraciones óseas en el raquitismo son más evidentes en los extremos inferiores del radio y el cúbito. Los extremos diafisarios pierden sus contornos claros y netos, tienen forma de copa y muestran una rarefacción moteada o en bandas. Posteriormente, la distancia entre los extremos del radio y el cúbito y los huesos metacarpianos aparece aumentada porque los verdaderos extremos no están calcificados y no se ven. Las sombras proyectadas por el tallo diafisario disminuyen de densidad, y la red formada por las laminillas se vuelve gruesa. Las deformaciones características se deben al incurvamiento de los huesos en la unión entre el cartílago y la diáfisis, ya que el tallo está debilitado. Cuando se inicia la curación, aparece en la epífisis una línea de calcificación blanca y delgada, que se hace más densa y gruesa a medida que avanza la calcificación. Más tarde se depositan sales de calcio por debajo del periostio, el tallo proyecta una sombra más densa y las laminillas desaparecen.
En los adultos, la desmineralización (osteomalacia) ocurre especialmente en la columna vertebral, la pelvis y las extremidades inferiores; las laminillas fibrosas se hacen visibles en las radiografías y en el córtex aparecen áreas de desmineralización incompleta en forma de cintas (seudofracturas, líneas de Looser, síndrome de Milkman). A medida que los huesos se ablandan, el peso puede causar arqueamiento de los huesos largos, acortamiento vertical de las vértebras y aplanamiento de la pelvis, lo cual estrecha la salida pélvica.
Datos de laboratorio
Pueden medirse en el plasma el 25(OH)D3 y otros metabolitos de la vitamina D. En personas sanas los niveles son 25 a 40 ng/ml (62,4 a 99,8 nmol/l) para el 25(OH)D3, y de 20 a 45 pg/ml (48 a 108 pmol/l) para la 1,25(OH)2D3. En el raquitismo nutricional y la osteomalacia, los niveles de 25(OH)D3 son muy bajos, y la 1,25(OH)2D3 es indetectable. Son característicos un fósforo sérico bajo (normal: 3 a 4,5 mg/dl [0,97 a 1,45 m mol/l]) y una fosfatasa alcalina sérica alta. El calcio sérico es bajo o normal, en función de la eficacia del hiperparatiroidismo secundario para restablecer el calcio sérico a la normalidad. La PTH sérica está elevada, y el calcio urinario está bajo en todas las formas de enfermedad, excepto en las que se asocian con acidosis. En el raquitismo hereditario dependiente de vitamina D, los datos de laboratorio varían
(v. más adelante).
Diagnóstico
Una historia de ingesta insuficiente de vitamina D orienta hacia el raquitismo y ayuda a distinguirlo del escorbuto infantil y de otras enfermedades. Puede distinguirse de la sífilis congénita (que se identifica mediante la serología y otras pruebas) y de la condrodistrofia (que se identifica por una cabeza grande, extremidades cortas, huesos gruesos y valores normales en suero de calcio, fósforo y fosfatasas).
La osteogénesis imperfecta, el cretinismo, la luxación congénita de cadera, la hidrocefalia y la poliomielitis se distinguirán fácilmente. Hay que diferenciar la tetania manifiesta en el raquitismo infantil de las convulsiones debidas a otras causas. El raquitismo refractario a la vitamina D puede ser causado por daño renal grave, o aparecer por acidosis tubular renal, hipofosfatemia familiar ligada al cromosoma X o al síndrome de Fanconi (v. Anomalías del transporte renal, cap. 261).
Hay que diferenciar la osteomalacia de otras causas de descalcificación ósea generalizada (p. ej., hiperparatiroidismo, osteoporosis senil o posmenopáusica, osteoporosis del hipertiroidismo, síndrome de Cushing, mieloma múltiple y atrofia por inactividad). Los cambios en los niveles séricos de calcio, fosfato, fosfatasa alcalina y 25(OH)D3, junto con los hallazgos radiológicos, confirman el diagnóstico.
Profilaxis
Se debe proporcionar una educación sanitaria a las poblaciones susceptibles, que incluya el consejo dietético. La leche humana es deficiente en vitamina D y contiene en promedio sólo 1,0 mg/l (40 UI/l), principalmente en forma de 25(OH)D3, mientras que la leche de vaca enriquecida contiene 10 mg/l (400 UI/l). A los lactantes criados con lactancia natural se les debe dar un suplemento de vitamina D de 7,5 mg (300 UI)/día desde el nacimiento hasta los 6 meses, en cuyo momento se dispone de una dieta más diversificada. El enriquecimiento con vitamina D de la harina de chapati sin levadura (125 mg/kg) ha sido eficaz entre los emigrantes hindúes en Gran Bretaña. Entre los adolescentes del Extremo Oriente, una sola dosis i.m. de 2,5 mg (100.000 UI) de ergocalciferol, administrada en el otoño, ha producido un considerable aumento de 25(OH)D3 en plasma que dura hasta la primavera.
Tratamiento
Con una ingesta suficiente de calcio y fósforo, la osteomalacia y el raquitismo no complicado pueden curarse administrando vitamina D en dosis de 40 mg (1.600 UI)/día. Los niveles séricos de 25(OH)D3 y 1,25(OH)2D3 empiezan a aumentar en 1 o 2 días. El fósforo sérico aumenta aproximadamente a los 10 días. La respuesta de un niño raquítico al tratamiento con vitamina D se muestra en la figura 3-1. Durante la tercera semana pueden observarse radiográficamente los signos de depósito de calcio y fósforo en los tejidos óseos. Aproximadamente 1 mes después del tratamiento se puede reducir la dosis gradualmente hasta el nivel de mantenimiento usual de 10 mg (400 UI)/día. Si hay tetania, la vitamina D debe suplementarse con sales de calcio i.v. durante la primera semana (v. Hipocalcemia en Alteraciones del metabolismo del calcio, cap. 12).
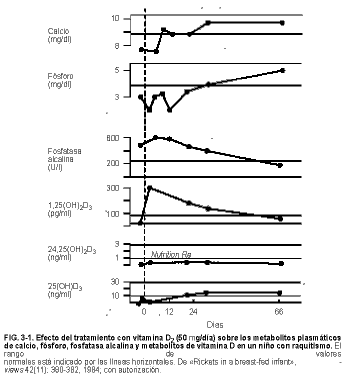
El raquitismo y la osteomalacia causados por defecto de la producción de metabolitos de la vitamina D (v. tabla 3-2) no responden a las dosis habituales eficaces en el raquitismo nutricional. Algunos casos responden a dosis masivas (600 a 1.200 mg de vitamina D2 o D3 diarias), pero puede producirse toxicidad. Cuando hay un defecto de producción de 25(OH)D3, las dosis de 50 mg/día de 25(OH)D3 elevan los niveles plasmáticos y producen una mejoría clínica.
RAQUITISMO HEREDITARIO DEPENDIENTE DE VITAMINA D
El tipo I (seudodeficiencia de vitamina D) es un síndrome autosómico recesivo caracterizado por un raquitismo grave, niveles plasmáticos normales de 25(OH)D3 y bajos de 1,25(OH)2D3, calcio sérico bajo o normal, hipofosfatemia y aminoaciduria generalizada. Este trastorno es consecuencia de 1a-hidroxilasa ausente o defectuosa en los riñones y responde a cantidades fisiológicas de 1,25(OH)2D3 (1 a 25 mg/día) i.v. u oral.
El tipo II existe en varias formas y se debe a mutaciones en el receptor de 1,25(OH)2D3. Este receptor, que es un factor de transcripción para 1,25(OH)2D3, causa la expresión de diversos genes que controlan el metabolismo de intestino, riñón, hueso y otras células. La ausencia de un receptor funcional produce un nivel alto pero ineficaz de 1,25(OH)2D3. Algunos Pacientes responden a dosis muy altas de 1,25(OH)2D3 (10 a 40 mg/día); otros no responden en absoluto.
TOXICIDAD DE VITAMINA D
Dosis de vitamina D de 1.000 mg (40.000 UI)/ día producen toxicidad en 1 a 4 meses en los lactantes, y dosis tan pequeñas como 75 mg (3.000 UI)/día pueden causar toxicidad con el transcurso de los años. Se han producido efectos tóxicos en adultos que recibían 2.500 mg (100.000 UI)/día durante varios meses. Elevados niveles de calcio sérico de 12 a 16 mg/dl (3 a 4 m mol/l) son un hallazgo constante cuando aparecen síntomas tóxicos; los niveles normales son de 8,5 a 10,5 mg/dl (2,12 a 2,62 m mol/l). En todos los Pacientes que reciben grandes dosis de vitamina D se debería medir el calcio sérico con frecuencia (al principio semanalmente, después mensualmente).
Los primeros síntomas son anorexia, náuseas y vómitos, seguidos de poliuria, polidipsia, debilidad, nerviosismo y prurito. La función renal se deteriora, como evidencian la baja densidad urinaria, la proteinuria, los cilindros y la azotemia. Pueden presentarse calcificaciones metastásicas, particularmente en los riñones. Los niveles plasmáticos de 25(OH)D3 están elevados hasta quince veces en la toxicidad por la vitamina D, mientras que los de 1,25(OH)2D3 suelen estar dentro del rango normal.
Una historia de ingesta excesiva de vitamina D es crítica para diferenciar esta patología de todos los demás estados hipercalcémicos. La toxicidad por vitamina D se produce comúnmente durante el tratamiento del hipoparatiroidismo (v. Hipocalcemia en Alteraciones del metabolismo del calcio, cap. 12) y con el uso equivocado de dosis masivas de vitaminas. En Gran Bretaña se ha observado la llamada hipercalcemia de la infancia con falta de crecimiento con una ingesta diaria de vitamina D de 50 a 75 mg (2.000 a 3.000 UI).
El síndrome de Williams consiste en hipercalcemia transitoria en la infancia con la tríada de estenosis aórtica supravalvular, retraso mental y facies de duendecillo. Los niveles de 1,25(OH)2D3 durante la fase de hipercalcemia son de 8 a 10 veces los normales. La mayoría de los casos se deben a un defecto no identificado en el metabolismo de la vitamina D más que a una ingesta excesiva.
La 1,25(OH)2D3 es 100 veces más potente que la vitamina D3. Cuando se emplea para tratar diversas enfermedades, es preciso vigilar los posibles efectos tóxicos del tratamiento prolongado.
El tratamiento consiste en suspender la vitamina, proporcionar una dieta pobre en calcio, mantener la orina ácida y administrar corticosteroides. Si se han producido lesión renal o calcificaciones metastásicas renales, éstas pueden ser irreversibles. Los diuréticos y la administración forzada de líquidos no tienen utilidad.
DEFICIENCIA DE VITAMINA E
Vitamina E es un término genérico para designar compuestos que tienen un anillo 6-cromanol, una cadena lateral isoprenoide y la actividad biológica del a-tocoferol. El grupo de la vitamina E incluye a los tocoferoles a, ß, g y a los d-tocoferoles, los cuales varían en la medida en que el anillo del cromanol está metilado. El d-a-tocoferol es el único esteroisómero presente en la naturaleza y el más potente en los ensayos biológicos (1,49 UI/mg); el dl-a-tocoferol, totalmente sintético, está racemizado completamente y tiene menos actividad biológica (1,1 UI/mg) que el d-a-tocoferol. El estándar internacional es el acetato de dl-a-tocoferol (1,0 UI/mg). En general, los tocoferoles actúan como antioxidantes, evitando la peroxidación de los lípidos con ácidos grasos poliinsaturados en las membranas celulares. La actividad antioxidante del a-tocoferol es similar a la de la glutatión peroxidasa, que contiene selenio (v. Selenio, cap. 4). Los niveles de tocoferol plasmáticos varían en los seres humanos con los niveles de lípidos plasmáticos totales, lo cual afecta a la partición entre el plasma y el tejido adiposo, el principal depósito de reserva para los tocoferoles. El nivel plasmático normal de a-tocoferol es de 5 a 10 mg/ml (11,6 a 23,2 m mol/l).
Las enfermedades causadas por la deficiencia de vitamina E varían considerablemente según las especies. La deficiencia puede causar alteraciones de la reproducción, anomalías de músculo, hígado, médula ósea y función cerebral, hemólisis de eritrocitos, embriogénesis defectuosa y diátesis exudativa, un trastorno de la permeabilidad capilar. Puede presentarse distrofia de músculo esquelético y, en ciertas especies, se acompaña de miocardiopatía.
En seres humanos, las principales manifestaciones de la deficiencia de vitamina E son: 1) anemia hemolítica leve asociada con aumento de hemólisis eritrocitaria y 2) enfermedad espinocerebelosa (v. en Trastornos cerebelosos y espinocerebelosos, cap. 179), la cual aparece sobre todo en niños que tienen malabsorción de grasas debida a abetalipoproteinemia, enfermedad hepatobiliar colestática crónica, enfermedad celíaca o una anomalía congénita en el metabolismo de la vitamina E.
La retinopatía de la prematuridad, también llamada fibroplasia retrolental (v. Retinopatía de la prematuridad, cap. 260), puede mejorar con el tratamiento con vitamina E, al igual que algunos casos de hemorragia intraventricular y subependimaria en el recién nacido.
Etiología
Los lactantes nacen en un estado de relativa deficiencia de vitamina E, con niveles plasmáticos de a-tocoferol inferiores a 5 mg/ml (11,6 m mol/l). El grado de deficiencia es tanto mayor cuanto más pequeño y más prematuro sea el lactante. La deficiencia de vitamina E en los lactantes prematuros persiste durante las primeras semanas de vida y puede atribuirse a transferencia placentaria limitada de vitamina E, bajos niveles tisulares al nacimiento, deficiencia dietética relativa en la infancia, malabsorción intestinal y crecimiento rápido. A medida que madura el aparato digestivo, la absorción de vitamina E mejora y los niveles de vitamina E en sangre aumentan.
La malabsorción subyace por lo general a la deficiencia de vitamina E en niños y adultos. También puede representar un papel una anomalía genética en el transporte de vitamina E.
Síntomas y signos
La anemia hemolítica en lactantes prematuros puede ser una manifestación de la deficiencia de vitamina E. En ese caso un lactante tiene niveles de hemoglobina de 7 a 9 g/dl, niveles bajos de vitamina E, reticulosis e hiperbilirrubinemia.
La abetalipoproteinemia (síndrome de Bassen-Kornzweig), debida a ausencia genética de apolipoproteína B, causa una intensa malabsorción de grasa y esteatorrea, con neuropatía progresiva y retinopatía en las primeras dos décadas de la vida (v. Abetalipoproteinemia, cap. 16). Los niveles de vitamina E suelen ser indetectables.
Los niños con enfermedad hepatobiliar colestática o fibrosis quística manifiestan el síndrome neurológico de deficiencia de vitamina E. Sus signos son ataxia espinocerebelosa con pérdida de reflejos tendinosos profundos, ataxia de tronco y extremidades, pérdida del sentido de la vibración y la postura, oftalmoplejía, debilidad muscular, ptosis palpebral y disartria. En adultos con malabsorción, la ataxia espinocerebelosa debida a deficiencia de vitamina E es sumamente rara, sin duda porque los adultos tienen grandes reservas de vitamina E en el tejido adiposo. Los rasgos clínicos de la deficiencia de vitamina E se enumeran en la tabla 3-3.
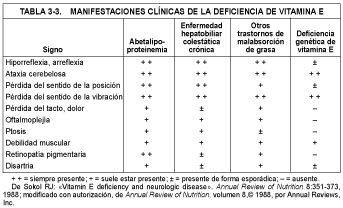
En una rara forma genética de deficiencia de vitamina E sin malabsorción de grasas, el hígado parece carecer de una proteína que normalmente transfiere d-a-tocoferol desde los hepatocitos a las lipoproteínas de muy baja densidad. Por ello no pueden mantenerse los niveles plasmáticos normales de a-tocoferol.
Datos de laboratorio y diagnóstico
Los lactantes prematuros deficientes en vitamina E tienen debilidad muscular, creatinuria y pigmentación ceroide con necrosis en las biopsias musculares. También se observa un aumento de hemólisis con el peróxido de hidrógeno. Los niveles plasmáticos de tocoferol son <4 mg/ml (<9,28 m mol/l). En los adultos, debe considerarse una deficiencia de vitamina E cuando el nivel plasmático de tocoferol es <5 mg/ml (<11,6 m mol/l) con un aumento de susceptibilidad de los eritrocitos al peróxido de hidrógeno. Si existe hiperlipidemia, el nivel de a-tocoferol está elevado, y la deficiencia se diagnostica cuando el nivel de tocoferol es <0,7 mg/g (<1,6 mmol/g) de grasa plasmática, lo cual corresponde a <5 mg/ml (<11,6 m mol/l) en una persona normolipémica.
En personas con deficiencia de vitamina E que toman una dieta exenta de creatina puede haber creatinuria excesiva y aumento de los niveles plasmáticos de creatinfosfocinasa. Pueden perderse los axones mielínicos de gran diámetro en la periferia, y los cordones posteriores de la médula espinal pueden degenerar en personas con enfermedad espinocerebelosa.
Tratamiento
La dosis profiláctica de a-tocoferol es de 0,5 mg/kg para lactantes nacidos a término y de 5 a 10 mg/kg para los lactantes prematuros.
En la malabsorción que causa deficiencia manifiesta, deben administrarse de 15 a 25 mg/kg/ día de a-tocoferol por v.o. en forma de d-a-tocoferilacetato hidrosoluble (1 mg = 1,4 UI).
Se necesitan dosis mucho mayores (hasta 100 mg/kg/día v.o. en dosis repartidas) para el tratamiento temprano de la neuropatía o para superar el defecto de absorción y transporte en la abetalipoproteinemia. Este tratamiento ha aliviado los síntomas en Pacientes jóvenes y ha detenido la neuropatía en Pacientes mayores.
En la forma genética de la deficiencia de vitamina E sin malabsorción de grasas, las dosis masivas de a-tocoferol (100 a 200 UI/día) mejoran la deficiencia y evitan las secuelas neurológicas.
TOXICIDAD DE VITAMINA E
Personas adultas han tomado cantidades relativamente grandes de vitamina E (400 a 800 mg/ día de d-a-tocoferol) durante meses o años sin ningún daño aparente. A veces se ha presentado debilidad muscular, fatiga, náuseas y diarrea en personas que toman de 800 a 3.200 mg/día. El efecto tóxico más importante de la vitamina E a dosis >1.000 mg/día es el antagonismo de la acción de la vitamina K y la potenciación del efecto de anticoagulantes cumarínicos orales, lo que puede conducir a hemorragia manifiesta.
DEFICIENCIA DE VITAMINA K
Vitamina K es un término genérico para los derivados de la 2-metil-1,4-naftoquinona que tienen actividad en la coagulación sanguínea. Las formas naturales están sustituidas en posición 3 con una cadena lateral alquílica. La vitamina K1 (filoquinona) tiene una cadena lateral de fitilo en posición 3 y es el único homólogo de la vitamina K que se encuentra en las plantas. La vitamina K2 designa a una familia de homólogos con 2-metil-1,4-naftoquinona sustituida en posición 3 con cadenas laterales de isoprenilo que contienen de 4 a 13 unidades de isopreno. Éstas se denominan menaquinonas; un sufijo (-n) indica el número de unidades de isopreno en la cadena lateral. Las menaquinonas son sintetizadas por bacterias en el tracto intestinal y pueden aportar una parte de las necesidades de vitamina K. La vitamina K es esencial porque el núcleo de la 1,4-naftoquinona no puede ser sintetizado en el organismo.
La vitamina K controla la formación en el hígado de los factores de la coagulación II (protrombina), VII (proconvertina), IX (factor Christmas, componente tromboplastínico del plasma)
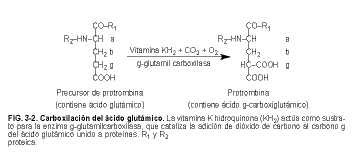
y X (factor Stuart). Otros factores de la coagulación dependientes de la vitamina K son la proteína C, la proteína S y la proteína Z; las proteínas C y S son anticoagulantes. Dos proteínas de la matriz ósea imprescindibles para el metabolismo óseo normal dependen de la vitamina K. Todas estas proteínas dependientes de la vitamina K contienen el aminoácido g-carboxiglutámico y todas ellas participan en reacciones que requieren calcio. La vitamina K participa en la conversión de 10-12 residuos de ácido glutámico en proteínas precursoras de la coagulación (p. ej., el precursor de la protrombina) a sus formas activas (p. ej., protrombina) mediante adición de dióxido de carbono (carboxilación; v. fig. 3-2). Esta adición incrementa la afinidad de los residuos de ácido glutámico por el calcio, lo que es esencial para la coagulación y para la modulación de la captación de calcio en el hueso.
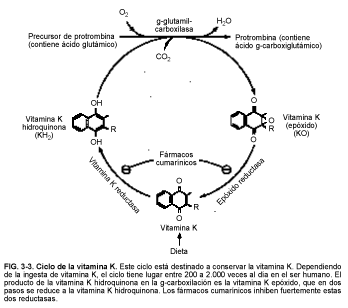
La figura 3-3 representa la reacción de carboxilación y el ciclo de la vitamina K, el cual es la vía de recuperación de la vitamina K. El epóxido de la vitamina K, el producto de vitamina K en la reacción de carboxilación del glutamilo, es reciclado a vitamina K hidroquinona mediante reducción enzimática. Los anticoagulantes cumarínicos no bloquean la reacción de carboxilación; en lugar de ello, inhiben a las dos reductasas que regeneran la vitamina K hidroquinona a partir del epóxido de vitamina K. La carboxilación está inhibida porque la vitamina K hidroquinona, un sustrato esencial para la g-glutamilcarboxilasa, no se ha formado. Las dosis grandes de vitamina K (1 a 10 mg) pueden superar el bloqueo cumarínico haciendo uso de otra reductasa en el hígado para regenerar la vitamina K hidroquinona.
Las necesidades diarias de vitamina K son alrededor de 1 mg/kg. La deficiencia de vitamina K causa hipoprotrombinemia y un descenso de la concentración de los demás factores de la coagulación dependientes de la vitamina K, y se manifiesta con coagulación defectuosa y hemorragia.
En los recién nacidos, el aporte nutricio de vitamina K no está asegurado porque: 1) la placenta transfiere los lípidos con relativa escasez; 2) el hígado del neonato es inmaduro con respecto a la síntesis de protrombina; 3) la leche materna es pobre en vitamina K, con un contenido de sólo 1 a 3 mg/l (la leche de vaca contiene 5 a 10 mg/l), y 4) el intestino del neonato es estéril durante los primeros días de vida. La enfermedad hemorrágica del recién nacido, causada por una deficiencia de vitamina K, ocurre generalmente de 1 a 7 días después del parto y puede manifestarse por hemorragia cutánea, GI, intratorácica o, en los casos peores, intracraneal. La enfermedad hemorrágica tardía, que tiene las mismas manifestaciones clínicas, ocurre de 1 a 3 meses después del parto. Suele asociarse con malabsorción o hepatopatía. Si la madre ha tomado anticonvulsivos, antibióticos cefalosporínicos o anticoagulantes cumarínicos, el riesgo de ambos tipos de enfermedad hemorrágica aumenta. La deficiencia de vitamina K en lactantes criados al pecho sigue siendo por ello una importante causa de morbididad y mortalidad en todo el mundo.
La deficiencia primaria de vitamina K es rara en los adultos sanos. Los adultos están protegidos frente a la carencia de vitamina K porque ésta está ampliamente distribuida en los tejidos de plantas y animales, el ciclo de la vitamina K conserva la vitamina y la flora microbiana del intestino normal sintetiza menaquinonas. Sin embargo, la deficiencia de vitamina K puede presentarse en adultos con una ingesta dietética marginal si experimentan traumatismos, cirugía extensa o nutrición parenteral con o sin tratamiento con antibióticos de amplio espectro. Las personas con obstrucción biliar, malabsorción o hepatopatía parenquimatosa también tienen un riesgo más elevado de deficiencia de vitamina K; son vulnerables a la enfermedad hemorrágica relacionada con la vitamina K los que toman ciertos fármacos, entre ellos anticonvulsivos, anticoagulantes, ciertos antibióticos (en especial las cefalosporinas), salicilatos y dosis masivas de vitamina A o E. Las personas que reciben warfarina deben intentar mantener constante la ingesta de vitamina K para evitar la fluctuación de los niveles de protrombina.
Síntomas y signos
Los síntomas y signos son causados por la hipoprotrombinemia y la depresión asociada de otros factores de la coagulación dependientes de vitamina K. El sangrado es la principal manifestación si la causa es una ingesta dietética insuficiente o un antagonismo de la vitamina K por fármacos. En la deficiencia de vitamina K se presenta una fácil formación de equimosis, sangrado de mucosas (especialmente epistaxis), hemorragia GI, menorragia y hematuria. Después de traumatismos puede rezumar sangre de los lugares de punción o las incisiones quirúrgicas, y en los lactantes puede presentarse una hemorragia intracraneal con amenaza de la vida. En la ictericia obstructiva, la hemorragia -cuando se produce- suele comenzar después del cuarto o quinto día. Puede iniciarse en forma de un lento rezumamiento de una herida quirúrgica, las encías, la nariz o la mucosa GI, o ser una hemorragia masiva en el tracto GI.
Datos de laboratorio
Una reducción en la actividad de la protrombina y otros factores de la coagulación dependientes de la vitamina K indica deficiencia o antagonismo de la vitamina K. El tiempo de protrombina (TP) y el tiempo de tromboplastina parcial activada (TTPa) suelen estar prolongados. El nivel de fibrinógeno, el tiempo de trombina, el recuento de plaquetas y el tiempo de hemorragia están en el intervalo normal. El nivel plasmático de filoquinona oscila entre 0,2 y 1,0 ng/ml en sujetos normales que consumen de 50 a 150 mg de filoquinona por día. La restricción de la ingesta de vitamina K a <50 mg/día rebaja el nivel plasmático. Sin embargo, medir los niveles plasmáticos sin conocer la ingesta de vitamina K no tiene utilidad en la detección selectiva de la deficiencia.
El indicador más sensible de la deficiencia de vitamina K es la presencia de des-g-carboxiprotrombina (DCP) en plasma. La DCP, también conocida como PIVKA (Protein Induced in Vitamin K Absence or Antagonism) (proteínas inducidas en ausencia o antagonismo de la vitamina K), puede medirse con los anticuerpos adecuados. Está ausente en el plasma de las personas sanas.
Diagnóstico
El diagnóstico se sospecha ante la presencia de los síntomas y los signos y una historia indicativa de la posibilidad de deficiencia de vitamina K, y se confirma cuando el TP y el TTPa están prolongados. Un ensayo terapéutico puede ayudar a excluir la patología hepática. Si se administra 1 mg de filoquinona solubilizada con ácidos grasos polioxietilados por vía i.v. (también conocida como filoquinona inyectable), y los niveles de protrombina aumentan significativamente en 2 a 6 horas, la hepatopatía es improbable. (Fitonadiona es el nombre genérico USP para las preparaciones de filoquinona utilizadas como fármaco, tanto inyectable como oral.) Otras enfermedades -como escorbuto, púrpura alérgica, leucemia y trombocitopenia, que pueden producir síntomas hemorrágicos- no se caracterizan por hipoprotrombinemia.
Profilaxis
Para los recién nacidos se recomienda rutinariamente de 0,5 a 1 mg i.m. para prevenir la hipoprotrombinemia y reducir la incidencia de hemorragia intracraneal por el traumatismo del parto. También se emplea profilácticamente cuando se contempla la posibilidad de una intervención quirúrgica. Otras alternativas son administrar fitonadiona a la madre en dosis profilácticas (2 a 5 mg/día v.o.) durante 1 semana antes de la fecha esperada del parto o solución de fitonadiona (2 a 5 mg i.m.) de 6 a 24 horas antes del parto. Las mujeres embarazadas que toman anticonvulsivos deben recibir fitonadiona a dosis de 20 mg/día v.o. 2 semanas antes del parto para evitar la hemorragia fetal. El bajo contenido de filoquinona en la leche materna no se debe a un consumo deficiente y no puede restablecerse comiendo diariamente vegetales frescos de hojas verdes.
Tratamiento
La filoquinona es el preparado de elección y se comercializa bajo el nombre genérico de fitonadiona. Puede utilizarse para tratar la hipoprotrombinemia, en particular la causada por los anticoagulantes derivados de la cumarina o la indanediona. El bisulfito sódico de menadiona es ineficaz contra estos antagonistas porque su conversión a 4-menaquinona es muy ineficiente (1%). Siempre que sea posible, la fitonadiona se debe administrar por vía s.c. o i.m. La dosis habitual en el adulto es de 10 mg i.m. En situaciones de urgencia, se deben administrar de 10 a 20 mg de fitonadiona inyectable disueltos en dextrosa al 5% o cloruro sódico al 0,9% por vía i.v. a una velocidad que no exceda 1 mg/min. (Aun cuando la fitonadiona esté correctamente diluida y se administre con lentitud, han ocurrido raras veces reacciones graves semejantes a la hipersensibilidad o a la anafilaxia, incluido el shock o la parada respiratoria.) Se puede repetir la dosis 6 a 8 horas después si el TP no se ha acortado de forma satisfactoria. La respuesta suele ser detectable en 1 o 2 horas, y en la mayoría de los casos este tratamiento es eficaz en 3 a 6 horas. La fitonadiona a dosis de 5 a 20 mg está indicada para el control de la hipoprotrombinemia en casos no urgentes en Pacientes que toman anticoagulantes. Los efectos beneficiosos suelen ser evidentes en 6 a 10 horas.
TOXICIDAD DE VITAMINA K
La filoquinona (vitamina K1) no es tóxica a dosis 500 veces superiores a las CDR (0,5 mg/kg/ día). Pero la menadiona, un precursor de la vitamina K, tiene toxicidad mensurable resultante de su reacción con los grupos sulfhidrilo; puede causar anemia hemolítica, hiperbilirrubinemia y kernicterus en los lactantes. La menadiona no debe usarse para tratar la deficiencia de vitamina K.
DEFICIENCIA Y DEPENDENCIA DE TIAMINA
La coenzima pirofosfato de tiamina, la forma activa de la tiamina (vitamina B1), participa en el metabolismo de los hidratos de carbono a través de la decarboxilación de los a-cetoácidos. La tiamina también actúa como coenzima de la apoenzima transcetolasa en la vía de los monofosfatos de pentosa para la glucosa. La deficiencia produce el beriberi, que cursa con manifestaciones neurológicas periféricas, cerebrales, cardiovasculares y gastrointestinales.
Etiología
La deficiencia primaria de tiamina se produce por una ingesta insuficiente de tiamina, particularmente en personas que se alimentan de arroz muy refinado. Al moler el arroz se quita la cáscara, que contiene la mayor parte de la tiamina, pero la ebullición antes de quitar la cáscara dispersa la vitamina por todo el grano, evitando así que se pierda.
La deficiencia secundaria de tiamina se produce por aumento de las necesidades, como en el hipertiroidismo, la gestación, la lactancia y la fiebre; alteración de la absorción, como en las diarreas prolongadas, y alteración de la utilización, como en la hepatopatía grave. En el alcoholismo se produce una combinación de disminución de ingesta, alteración de la absorción y la utilización, mayores necesidades y, posiblemente, una deficiencia de apoenzima. Las infusiones de dextrosa frecuentes, prolongadas o muy concentradas, junto con una ingesta baja de tiamina, pueden desencadenar una deficiencia de esta última.
Anatomía patológica
Las alteraciones neurales más avanzadas se producen en los nervios periféricos, particularmente en las piernas. De modo característico se afectan primero y más gravemente los segmentos distales. Puede producirse degeneración de la vaina medular en todos los tractos de la médula espinal, especialmente en los cordones posteriores y en las raíces nerviosas anteriores y posteriores. También se producen cambios en el asta anterior y en las células ganglionares posteriores. Cuando la deficiencia es grave se presentan lesiones de polioencefalitis hemorrágica.
El corazón está dilatado y aumentado de tamaño; las fibras musculares hinchadas, fragmentadas y vacuolizadas, con esPacios intersticiales dilatados por acumulación de líquido. Se produce vasodilatación y puede conducir a una pequeña cantidad de edema antes de llegar a la insuficiencia cardíaca con gran volumen de eyección.
Síntomas y signos
La deficiencia en una etapa temprana produce fatiga, irritabilidad, pérdida de la memoria, alteraciones del sueño, dolor precordial, anorexia, malestar abdominal y estreñimiento.
El síndrome de alteraciones neurológicas periféricas debido a deficiencia de tiamina se llama beriberi seco. Esas alteraciones son bilaterales y simétricas, predominan en las extremidades inferiores y se inician con parestesias de los dedos de los pies, sensación de quemazón en los pies (particularmente importante durante la noche), calambres musculares en las pantorrillas y dolores en las piernas. El dolorimiento de los músculos de la pantorrilla, la dificultad para ponerse de pie desde la posición en cuclillas, una disminución de la sensación vibratoria en los dedos de los pies y la disestesia plantar son signos tempranos. Puede establecerse el diagnóstico de neuropatía periférica leve cuando los reflejos aquíleos están ausentes. La deficiencia continuada produce abolición del reflejo rotuliano, pérdida de la sensación vibratoria y posicional de los dedos de los pies, atrofia de los músculos de la pantorrilla y del muslo, y finalmente pie y dedos péndulos. Una vez que los signos de las piernas están bien establecidos pueden afectarse los brazos.
El beriberi cerebral (síndrome de Wernicke-Korsakoff) se produce por una deficiencia aguda grave superpuesta a la deficiencia crónica (v. Amnesias, cap. 169). La etapa inicial, llamada síndrome de Korsakoff, se caracteriza por confusión mental, afonía y confabulación. El flujo sanguíneo cerebral está notablemente disminuido y la resistencia vascular aumentada. La encefalopatía de Wernicke consiste en nistagmo, oftalmoplejía total, coma y muerte en los casos no tratados.
El beriberi cardiovascular (húmedo) (beriberi Shoshin) se presenta en la deficiencia de tiamina cuando destaca la enfermedad miocárdica. Ésta origina un gasto cardíaco elevado con vasodilatación y extremidades calientes. Antes de que se produzca la insuficiencia cardíaca, aparece taquicardia, presión del pulso amplia, sudoración y piel caliente, y se desarrolla acidosis láctica. Con el fallo cardíaco se presentan ortopnea y edema pulmonar y periférico; y la vasodilatación continúa, llegando a veces al shock.
El beriberi infantil se produce en lactantes (habitualmente entre el segundo y cuarto mes de vida) que son alimentados con lactancia natural por madres con deficiencia de tiamina. Son características la insuficiencia cardíaca, la afonía y la ausencia de reflejos tendinosos profundos.
Datos de laboratorio
El aumento de piruvato y lactato en sangre y la disminución de la excreción urinaria de tiamina (<50 mg/día) son compatibles con el diagnóstico de deficiencia de tiamina. La actividad de la transcetolasa eritrocitaria es menor antes y mayor tras la administración de pirofosfato de tiamina (efecto PPT) y es un indicador sensible de reservas tisulares. Las variaciones de niveles de apoenzimas en algunos casos pueden complicar la interpretación de la prueba.
Diagnóstico
En la diabetes mellitus no controlada o de larga duración y en el alcoholismo se produce una forma de polineuropatía que no responde a la tiamina y que es clínicamente similar a la de su deficiencia. Otras formas de polineuropatía simétrica bilateral de comienzo en las piernas son raras. Es poco probable que se deban a la deficiencia de tiamina las mononeuritis (mononeuropatías), como la ciática, y las polineuropatías que se inician en otras partes del cuerpo.
El diagnóstico del beriberi cardiovascular es difícil cuando la deficiencia de tiamina está complicada por cardiopatía hipertensiva o degenerativa, miocardiopatía viral o fiebre reumática. La respuesta terapéutica a la tiamina puede ser útil para establecer el diagnóstico.
Tratamiento
En la polineuropatía leve se administran de 10 a 20 mg/día de tiamina en dosis fraccionadas durante 2 semanas, seguidas de una dieta nutritiva. La dosificación es de 20 a 30 mg/día en la neuropatía moderada o avanzada y debe continuar durante varias semanas tras la desaparición de los síntomas. El edema y la congestión del beriberi Shoshin responden en pocas horas a 100 mg/día de tiamina i.v., lo que debe continuar durante varios días, además del reposo en cama. La insuficiencia cardíaca debida al beriberi responde escasamente a la digital y a los diuréticos.
En el síndrome de Wernicke-Korsakoff suele ser preciso administrar de 50 a 100 mg de tiamina i.m. o i.v. dos veces al día durante varios días, seguidos por 10 a 20 mg diarios hasta que se obtenga una respuesta terapéutica. Son raras las reacciones anafilácticas a la tiamina i.v. y no tienen relación con la dosis.
La deficiencia de tiamina no se asocia en general a otras deficiencias de complejo B, y suele ser aconsejable el tratamiento con múltiples vitaminas hidrosolubles a dosis 5 a 10 veces superiores a las CDR en varias semanas. Este régimen debe ir seguido indefinidamente por una dieta nutritiva que aporte dosis de una a dos veces las CDR.
El magnesio, un cofactor de la transcetolasa, debe administrarse en forma de sulfato de magnesio (1 a 2 ml i.m. de una solución al 50%) con tiamina para corregir la resistencia a ésta y la hipomagnesemia que la acompaña con frecuencia. La hiponatremia (cap. 12) debe corregirse lentamente, porque una corrección rápida puede causar mielinólisis pontina central. La recuperación de los problemas neurológicos suele ser incompleta en el beriberi. En el beriberi cerebral, la mielinólisis pontina central puede ser residual.
Dependencia de tiamina: Varios defectos congénitos del metabolismo responden a dosis farmacológicas de tiamina (5 a 20 mg/día). Incluyen una anemia megaloblástica de origen desconocido, la acidosis láctica debida a baja actividad de la piruvato deshidrogenasa hepática y la cetoaciduria debida a baja actividad de las deshidrogenasas de los cetoácidos ramificados.
DEFICIENCIA DE RIBOFLAVINA
La riboflavina (vitamina B2), en forma de flavina mononucleótido o de flavina adenina dinucleótido, actúa como una coenzima esencial en muchas reacciones de oxidación-reducción involucradas en el metabolismo de los hidratos de carbono. La deficiencia conduce a lesiones orales, oculares, cutáneas y genitales.
La deficiencia primaria de riboflavina está asociada con el consumo insuficiente de leche y otros productos animales. Las deficiencias secundarias son más comunes en diarreas crónicas, hepatopatías, alcoholismo crónico y situaciones postoperatorias en las cuales las infusiones de nutrientes carecen de vitaminas suplementarias o síntomas.
Síntomas, signos y datos de laboratorio
Los signos más frecuentes son palidez y maceración de la mucosa en las comisuras de la boca (estomatitis angular) y las superficies rojas de los labios (queilosis), seguida de fisuras lineales superficiales que pueden dejar cicatrices al curar. Cuando esas lesiones se infectan con Candida albicans, se producen lesiones exuberantes de color blanco grisáceo (boqueras). La lengua puede tener un aspecto violáceo. Las lesiones cutáneas suelen afectar a los pliegues nasolabiales, las alas de la nariz, las orejas, los párpados, el escroto y los labios mayores. Estas áreas se vuelven rojas, escamosas y grasientas, y se acumula material sebáceo en los folículos pilosos, produciendo seborrea o piel de tiburón. Raras veces aparece neovascularización de la córnea y queratitis epitelial, con lagrimeo y fotofobia. La ambliopía funcional puede responder a la riboflavina.
Una excreción urinaria <30 mg de riboflavina/g de creatinina se asocia con signos clínicos de deficiencia de riboflavina. El aumento de activación de la glutatión reductasa eritrocitaria por la riboflavina es un signo temprano de deficiencia.
Diagnóstico y tratamiento
Las lesiones descritas no se presentan solamente en la deficiencia de riboflavina. La queilosis puede ser consecuencia de deficiencia de vitamina B6, ausencia de dientes o dentaduras mal ajustadas. La dermatitis seborreica y las lesiones oculares pueden producirse en diversas patologías. Por tanto, el diagnóstico de la deficiencia de riboflavina no puede depender sólo de la historia y la presencia de lesiones sospechosas. Pueden ser necesarias pruebas de laboratorio, la eliminación de otras causas y un ensayo terapéutico.
Se administran de 10 a 30 mg/d v.o. de riboflavina en dosis fraccionadas hasta que se observa una respuesta, y después de 2 a 4 mg/d hasta la recuperación. La riboflavina se puede administrar por vía i.m. a razón de 5 a 20 mg/d en dosis única o fraccionada.
DEFICIENCIA DE NIACINA
Los derivados de la niacina (ácido nicotínico) son el nicotinamida-adenina dinucleótido (NAD, coenzima I) y el nicotinamida-adenina dinucleótido fosfato (NADP, coenzima II), que son coenzimas en reacciones de oxidación-reducción. Son vitales en el metabolismo celular.
Etiología
Deficiencias graves de niacina y triptófano, un precursor a partir del cual el organismo puede sintetizar niacina, son las principales causas de la pelagra. La deficiencia primaria suele presentarse en áreas geográficas donde el maíz constituye la parte principal de la dieta. La niacina ligada, que se encuentra en el maíz, no se asimila en el tracto intestinal a menos que haya sido tratada previamente con álcalis, como en la preparación de tortitas. La proteína del trigo también es deficiente en triptófano. La deficiencia puede contribuir además al desequilibrio de los aminoácidos, puesto que la pelagra es frecuente en la India entre las personas que comen un mijo con un contenido alto de leucina.
La deficiencia secundaria se observa en diarreas, cirrosis y alcoholismo y asimismo tras el uso postoperatorio amplio de infusiones de nutrientes que carecen de vitaminas. La pelagra puede aparecer durante el tratamiento prolongado con isoniazida (el fármaco reemplaza a la niacinamida en el NAD), en el tumor carcinoide maligno (el triptófano es desviado para formar 5-hidroxitriptamina) y en la enfermedad de Hartnup (v. Anomalías del transporte renal, cap. 261).
Síntomas y signos
La pelagra se caracteriza por síntomas cutáneos, de las membranas mucosas, del SNC y GI. El síndrome completo de deficiencia avanzada incluye erupción fotosensitiva simétrica, estomatitis escarlata, glositis, diarrea y alteraciones mentales. Los síntomas pueden presentarse solos o combinados.
Se han identificado cuatro tipos de lesiones cutáneas, generalmente bilaterales y simétricas:
• Lesiones agudas que consisten en eritema seguido de vesiculación, ampollas, costras y descamación; la infección secundaria es frecuente, sobre todo tras la exposición a la luz solar (traumatismo actínico).
• Intértrigo, también agudo, caracterizado por enrojecimiento, maceración, abrasión e infección secundaria en las áreas intertriginosas.
• Hipertrofia crónica, en la cual la piel está engrosada, sin elasticidad, fisurada e intensamente pigmentada sobre los puntos de presión; con frecuencia se presenta infección secundaria, y la lesión tiene un borde perlado netamente definido de epitelio en regeneración cuando se inicia la curación.
• Lesiones atróficas crónicas, con piel seca, descamativa, sin elasticidad y demasiado grande para la parte que cubre (lo que se observa en las pelagras de los ancianos).
La distribución de las lesiones, en los puntos con traumatismos, es más característica que su forma. La luz solar origina el collar de Casal y las lesiones de la cara en forma de mariposa.
Los síntomas de las membranas mucosas afectan principalmente a la boca, pero también pueden implicar a la vagina y la uretra. La glositis y la estomatitis escarlata son características de la deficiencia aguda. La punta y los bordes de la lengua y la mucosa que rodea el conducto de Stenon se afectan en primer lugar. A medida que la lesión progresa, la totalidad de la lengua y las mucosas orales adquieren un color escarlata brillante, seguido de dolor bucal, aumento de salivación y edema de la lengua. Pueden aparecer ulceraciones bajo la lengua, sobre la mucosa del labio inferior y frente a los molares. Suelen estar cubiertas con un esfacelo grisáceo que contiene microorganismos de Vincent.
Entre los síntomas GI, que son imprecisos en los casos tempranos, hay sensación de quemazón en la boca, la faringe y el esófago y molestias y distensión abdominales. Más tarde pueden presentarse naúseas, vómitos y diarrea. La diarrea, frecuentemente sanguinolenta por la hiperemia del tracto GI y por la ulceración, es grave.
Los síntomas del SNC incluyen: 1) psicosis orgánica, caracterizada por deterioro de la memoria, desorientación, confusión y confabulación (excitación, depresión, manía y delirio predominan en algunos Pacientes; en otros, la reacción es paranoide), y 2) síndrome encefalopático, caracterizado por obnubilación de la conciencia, rigidez en rueda dentada de las extremidades y reflejos de succión y de prensión incontrolables. Es difícil diferenciar estas alteraciones del SNC de las de la deficiencia de tiamina.
Diagnóstico y tratamiento
Es preciso distinguir la deficiencia de niacina de las demás causas de estomatitis, glositis, diarrea y demencia. El diagnóstico es fácil cuando los hallazgos clínicos comprenden lesiones de la piel y la boca, diarrea, delirio y demencia. Lo más frecuente es que el trastorno no esté plenamente desarrollado, y tiene importancia una historia de dieta carente de niacina y triptófano. La excreción urinaria de N'-metilnicotinamida (NMN) y de su piridona está disminuida. Una excreción de NMN <0,8 mg/día indica una deficiencia de niacina.
Las deficiencias múltiples de vitaminas B y proteínas suelen presentarse juntas; por ello es necesaria una dieta equilibrada. Los suplementos de niacina, de 300 a 1.000 mg/día, deben administrarse por v.o. en dosis fraccionadas. En la mayoría de los casos es suficiente con 300 a 500 mg. Para tratar los estados de deficiencia se usa generalmente la niacinamida, porque la niacina puede causar enrojecimiento, prurito, ardor o sensaciones de hormigueo, lo que no ocurre con la niacinamida; sin embargo, la niacinamida no posee las propiedades hipolipemiantes o vasodilatadoras de la niacina. Cuando hay que evitar el tratamiento oral debido a diarrea o falta de cooperación del Paciente, deben inyectarse de 100 a 250 mg s.c. dos o tres veces al día. En los estados encefalopáticos se recomiendan 1.000 mg v.o. más 100 a 250 mg i.m. También deben administrarse otras vitaminas del complejo B en dosis terapéuticas.
DEFICIENCIA Y DEPENDENCIA DE VITAMINA B6
La vitamina B6 constituye un grupo de compuestos estrechamente emparentados: piridoxina, piridoxal y piridoxamina. Son metabolizados y fosforilados en el organismo a fosfato de piridoxal, el cual actúa como coenzima en muchas reacciones, incluidas la decarboxilación y la transaminación de aminoácidos, la desaminación de hidroxiaminoácidos y de cisteína, la conversión de triptófano a niacina y el metabolismo de los ácidos grasos. En consecuencia, el grupo de la vitamina B6 es importante en la sangre, el SNC y el metabolismo de la piel. La vitamina B6 es importante en la eritropoyesis porque el fosfato de piridoxal es imprescindible en la formación de ácido d-aminolevulínico, el paso limitante en la biosíntesis del hemo.
La deficiencia primaria es rara, porque la mayoría de los alimentos contienen vitamina B6. A pesar de todo, se ha observado un brote de convulsiones en lactantes consecutivo a la destrucción involuntaria de vitamina B6 en las fórmulas de leche artificial. La deficiencia secundaria puede ser consecuencia de malabsorción, alcoholismo, uso de anticonceptivos orales, inactivación química de fármacos (p. ej., hidracida del ácido isonicotínico, cicloserina, hidralazina, penicilamina), pérdida excesiva y aumento de la actividad metabólica.
Síntomas y signos
Deficiencia: El antagonista de la vitamina B6, la desoxipiridoxina, produce dermatitis seborreica, glositis, queilosis, neuropatía periférica y linfopenia. La deficiencia de vitamina B6 puede causar convulsiones en los lactantes y anemia en los adultos (generalmente normocítica pero a veces microcítica).
Dependencia: Varios estados recesivos ligados al cromosoma X afectan a diferentes apoenzimas de la vitamina B6, y producen síntomas como convulsiones, deficiencia mental, cistationinuria, anemia sideroblástica (por sobrecarga de hierro), urticaria, asma y aciduria xanturénica.
Datos de laboratorio y diagnóstico
Hasta el momento no existe una prueba generalmente aceptada del estado de la vitamina B6. El nivel de fosfato de piridoxal en sangre total es un indicador mejor que el nivel plasmático. Las actividades de las transaminasas glutámico-pirúvica y glutámico-oxalacética eritrocitarias están disminuidas en la deficiencia de vitamina B6, pero estos cambios no son diagnósticos a causa del amplio rango de valores en personas sanas.
Tratamiento
Deben corregirse las causas subyacentes, como el uso de fármacos inactivadores de la piridoxina (anticonvulsivos, corticosteroides, estrógenos, isoniazida, penicilamina e hidralazina) o la malabsorción. Para la dependencia en los lactantes, las necesidades diarias (normalmente 0,4 mg) están aumentadas muchas veces (hasta 10 mg). En caso de convulsiones dependientes de piridoxina, la dosis inicial es de 50 a 100 mg i.m. o i.v. diariamente en 1 semana seguida de dosis orales que se disminuyen progresivamente hasta 25 mg durante 1 semana. La deficiencia en los adultos suele responder a la piridoxina en dosis de 50 a 100 mg/día v.o. Las enfermedades que aumentan la demanda metabólica, como hipertiroidismo y diabetes, exigen cantidades mayores que las recomendadas. En la deficiencia de piridoxina asociada con fármacos como la isoniazida pueden necesitarse 100 mg/día. Para la dependencia en los adultos, pueden ser imprescindibles cantidades diarias tan grandes como 200 a 600 mg de piridoxina.
TOXICIDAD DE VITAMINA B6
La ingestión de dosis masivas (2 a 6 g/día durante 2 a 40 meses) de piridoxina, tomadas erróneamente para la tensión premenstrual, puede causar ataxia sensitiva progresiva y alteración del sentido de la posición y las vibraciones en las extremidades inferiores. Los sentidos del tacto, la temperatura y el dolor están menos afectados. No se deteriora el sistema motor ni el sistema nervioso central. La recuperación es lenta y, en algunos Pacientes, sólo es parcial tras la supresión de la ingestión de piridoxina.
DEFICIENCIA Y DEPENDENCIA DE BIOTINA
La biotina actúa como coenzima en la transferencia de dióxido de carbono y, por tanto, es esencial para el metabolismo de las grasas y los hidratos de carbono. Una enzima específica une la biotina a sus apoenzimas.
Deficiencia: La clara de huevo cruda contiene un antagonista de la biotina, la avidina. El consumo prolongado de claras de huevo crudas puede conducir a dermatitis y glositis, que responden con rapidez a la administración diaria de 150 a 300 mg de biotina. La deficiencia se ha presentado también en la NPT prolongada sin suplementos de biotina.
Dependencia: Se han descrito retraso del desarrollo físico y mental, alopecia, queratoconjuntivitis y defectos en la inmunidad por células T y células B en niños con deficiencias múltiples de carboxilasas dependientes de biotina. Las deficiencias pueden ser consecuencia de mutaciones en la holocarboxilasa sintetasa (enzima necesaria para unir la biotina a las cuatro carboxilasas imprescindibles para el metabolismo) o en la biotina de las mismas enzimas en el catabolismo). La excreción urinaria de varios ácidos orgánicos ayuda al diagnóstico. Los niños con anomalías de holocarboxilasa sintetasa y biotinidasa responden bien a grandes dosis de biotina (5 a 20 mg) diarias.
DEFICIENCIA DE ÁCIDO PANTOTÉNICO
El ácido pantoténico es una vitamina ampliamente distribuida en los alimentos y es un componente esencial de la coenzima A, que actúa como cofactor en la transferencia de acilos para muchas reacciones enzimáticas. Los adultos necesitan probablemente unos 4 a 7 mg/día, que corresponden a un nivel en sangre de 100 a 180 mg/dl (4,56 a 8,21 m mol/l), pero no se han establecido las CRD. La deficiencia de ácido pantoténico se observa rara vez en seres humanos.
Adultos voluntarios sometidos a una dieta deficiente experimentaron malestar, molestias abdominales y quemazón en los pies asociada con parestesias, que respondieron al ácido pantoténico. Pero estos síntomas inespecíficos responden raras veces en la práctica clínica a la vitamina.
DEFICIENCIA DE VITAMINA C
La vitamina C (ácido ascórbico) es esencial para la formación de colágeno y ayuda a mantener la integridad de las sustancias de origen mesenquimatoso, como el tejido conjuntivo, el osteoide tisular y la dentina. Es esencial para la cicatrización de las heridas y facilita la recuperación de las quemaduras. Esta vitamina es un potente agente reductor y es oxidada y reducida reversiblemente en el organismo, funcionando como un sistema redox en la célula. Está implicada en el metabolismo de la fenilalanina y la tirosina. Como agente reductor (con el oxígeno, el ion ferroso y un 2-cetoácido), la vitamina C activa las enzimas que hidroxilan la prolina y la lisina del procolágeno a hidroxiprolina e hidroxilisina del protocolágeno. En los animales escorbúticos, la elastina se hace cada vez más deficiente en hidroxiprolina. La vitamina C protege a la reductasa del ácido fólico, que convierte el ácido fólico a ácido folínico, y puede ayudar a la liberación de ácido fólico libre a partir de sus formas conjugadas en los alimentos. La vitamina C facilita la absorción de hierro. La deficiencia grave produce el escorbuto, una enfermedad crónica con manifestaciones hemorrágicas y formación de osteoide y dentina anormales.
Etiología
En adultos, la deficiencia primaria suele deberse a idiosincrasias alimentarias o a dietas inadecuadas. Las deficiencias se presentan en las enfermedades GI, en especial cuando el Paciente está a «régimen de úlcera». Embarazo, lactancia y tirotoxicosis aumentan las necesidades de vitamina C; las enfermedades inflamatorias agudas y crónicas, la cirugía y las quemaduras aumentan las necesidades de manera importante. La diarrea incrementa las pérdidas fecales y la aclorhidria reduce la cantidad absorbida. El estrés por frío o calor aumenta la excreción urinaria de vitamina C. El calor (p. ej., esterilización de fórmulas de alimentación artificial, cocinado) puede destruir la vitamina C de los alimentos.
Anatomía patológica
La formación de las sustancias del cemento intercelular en los tejidos conjuntivos, el hueso y la dentina es defectuosa, lo que origina capilares debilitados y subsiguiente hemorragia y defectos en el hueso y estructuras relacionadas. Las hemorragias están organizadas de forma avascular, de modo que las heridas cicatrizan mal y se abren con facilidad. El crecimiento endocondral se detiene porque los osteoblastos dejan de formar tejido osteoide, y se producen lesiones óseas. En su lugar se forma una unión fibrosa entre diáfisis y epífisis, y las uniones costocondrales aumentan de tamaño. En este tejido fibroso están englobados fragmentos de cartílago densamente calcificados. Las pequeñas hemorragias equimóticas que se producen dentro del hueso o a lo largo del mismo, o las grandes hemorragias subperiósticas debidas a pequeñas fracturas inmediatas a la línea blanca en dirección a la diáfisis, complican estas lesiones.
Síntomas y signos
En los adultos, el escorbuto permanece latente durante 3 a 6 meses tras la reducción de la vitamina C en la dieta a <10 mg/día. El escorbuto manifiesto va precedido por lasitud, debilidad, irritabilidad, pérdida de peso y vagas mialgias y artralgias. Las múltiples hemorragias subungueales pueden formar una media luna cerca del extremo distal de la uña y son más extensas que las de la endocarditis bacteriana. Las encías se vuelven hinchadas, de color púrpura, esponjosas y friables; en la deficiencia extrema sangran con facilidad. Con el tiempo se producen infecciones secundarias, gangrena y aflojamiento de los dientes. Estas alteraciones afectan solamente a la encía que rodea los dientes naturales o a los que tienen raíces ocultas. Las cicatrices antiguas se abren, las heridas nuevas no cicatrizan y pueden producirse hemorragias en cualquier parte del cuerpo, especialmente en forma de petequias perifoliculares y equimosis en la piel de los miembros inferiores. (En ancianos, estas alteraciones no son necesariamente escorbúticas.) En los adultos no se producen lesiones óseas, a excepción de la hemorragia subperióstica.
Otros síntomas y signos de escorbuto son la hemorragia de la conjuntiva bulbar, la neuropatía femoral por hemorragia en las vainas femorales, la oliguria, el edema de las extremidades inferiores, el deterioro de la reactividad vascular y la artritis parecida a la AR. Las encías sangrantes no son el rasgo más característico del escorbuto. El folículo piloso hiperqueratósico con hiperemia o hemorragia circundante es casi patognomónico.
Datos de laboratorio y diagnóstico
El ácido ascórbico plasmático cae desde el intervalo normal de 0,6 a 1,4 mg/dl (34 a 79 m mol/l) a <0,2 mg/dl (<11 m mol/l), a veces casi a cero. Los niveles de ácido ascórbico en la capa leucoplaquetaria de la sangre centrifugada son más significativos; los niveles normales >16 mg/108 células (>91 nmol/108 células) se reducen a <2,0 mg/108 células (<11,4 nmol/108 células). Cuando las reservas de vitamina C están agotadas, aparece poca cantidad en la orina tras una dosis de prueba de vitamina C. Una prueba de fragilidad capilar positiva es un hallazgo casi constante, y la anemia es frecuente. Los tiempos de hemorragia, coagulación y protrombina son normales.
En los adultos es preciso diferenciar el escorbuto de la artritis, las enfermedades hemorrágicas y la gingivitis. Los síntomas articulares se deben a sangrado alrededor de la articulación o en su interior. La presencia de hemorragias petequiales además de los estudios sanguíneos ayudan en el diagnóstico.
Profilaxis y tratamiento
Una dosis de vitamina C de 60 mg/día v.o. es completamente protectora. La mayoría de los especialistas en nutrición creen que las dosis enormes de vitamina C (unos 10 g/día) no reducen la incidencia o la gravedad del resfriado común
(v. Enfermedades virales respiratorias, cap. 162) ni influyen sobre la evolución de una enfermedad maligna o la arteriosclerosis. Estas dosis masivas acidifican la orina, pueden causar diarrea por los efectos osmóticos, predisponen a los cálculos urinarios de oxalato y promueven la sobrecarga de hierro.
Para el escorbuto en los adultos, se administran 100 mg de ácido ascórbico v.o. tres veces al día durante un período de 1 a 2 semanas, hasta la desaparición de los síntomas, seguidos por una dieta nutritiva que aporte una a dos veces la CDR. Después pueden administrarse las dosis de mantenimiento habituales. En los Pacientes escorbúticos, las dosis terapéuticas de ácido ascórbico restablecen las funciones de la vitamina C en pocos días. Los signos y los síntomas suelen desaparecer en 1 a 2 semanas. La gingivitis crónica con hemorragia subcutánea extensa puede tardar algo más en curar.