301 / FACTORES QUE MODIFICAN LA RESPUESTA FARMACOLÓGICA
FARMACOGENÉTICA
Una reacción farmacogenética es una variación en la respuesta farmacológica producida por factores hereditarios. A menudo, los parámetros cinéticos (p. ej., constante de Michaelis [Km], velocidad máxima de reacción [Vmax]) o las enzimas encargadas de metabolizar fármacos varían ampliamente entre sujetos normales. A veces, las tasas de eliminación varían de 4 a más de 40 veces, dependiendo del fármaco y la población estudiada. Los estudios que se han llevado a cabo en gemelos y en familias enteras han demostrado que existen factores genéticos que explican esta gran variabilidad interindividual.
La farmacogenética tiene, por tanto, un gran significado biológico e importantes consecuencias clínicas. Cuando se prescriben fármacos, los médicos deben tener en cuenta que la capacidad individual de aclaramiento de un fármaco puede variar mucho de un paciente a otro. Un paciente con metabolismo rápido requiere dosis más elevadas y más frecuentes para alcanzar concentraciones terapéuticas; un paciente con metabolismo lento puede necesitar dosis menores y menos frecuentes para evitar reacciones adversas, especialmente en caso de fármacos con estrecho margen de seguridad.
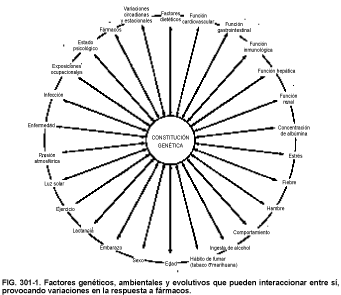
En la figura 301-1 se muestran los numerosos factores ambientales y evolutivos que pueden interactuar entre sí y con los factores genéticos, dando como resultado una alteración de la respuesta farmacológica. Un ejemplo muy frecuente es la edad: el paso del tiempo afecta a la mayoría de los factores, complicando la farmacocinética.
VARIACIONES FARMACOCINÉTICAS
Acetilación. Aproximadamente en el 50% de la población de Estados Unidos, la N-acetiltransferasa hepática es hipoactiva. Estas personas (acetiladores lentos) requieren más tiempo para metabolizar los fármacos que sufren procesos de acetilación y tienden a ser más susceptibles a los efectos indeseables asociados a estos fármacos (p. ej., neuritis periférica por isoniazida, lupus eritematoso por hidralazina o procainamida, sedación y náuseas por fenelzina).
En el resto de la población, la acetilación es rápida. En comparación con los acetiladores lentos, estas personas requieren mayores dosis o más frecuentes de fármacos que son acetilados (p.ej.,isoniazida) para obtener la respuesta terapéutica deseada. Los acetiladores rápidos, sin embargo, son más propensos a desarrollar hepatotoxicidad por la acumulación de hidralazina.
Hidrólisis. Las personas con déficit de seudocolinesterasa (aproximadamente 1:1.500) tienen una disminución de la capacidad de inactivar la succinilcolina, lo que desencadena una parálisis prolongada de los músculos respiratorios cuando reciben dosis convencionales del fármaco. La apnea prolongada puede requerir la aplicación de ventlación mecánica hasta que el fármaco pueda ser eliminado por otras vías.
Oxidación. Alrededor de un 5-10% de la población blanca de Norteamérica y Europa presenta una biotransformación lenta de la debrisoquina. Si alguna de estas personas toma debrisoquina como tratamiento de la hipertensión, con toda seguridad padecerá toxicidad (hipotensión ortostática). La hidroxilación reducida de diversos fármacos se ha correlacionado con una respuesta terapéutica inusualmente prolongada (p. ej., bloqueo prolongado de los receptores (con metoprolol o timolol) o una toxicidad superior a la esperada (p. ej., excesiva depresión del SNC con nortriptilina o difenilhidantoína). Los antidepresivos tricíclicos (amitriptilina, desipramina), algunos antiarrítmicos (encainida, flecainida) y un antitusígeno (el dextrometorfano) también pueden verse afectados por este déficit metabólico.
Cerca de un 4% de la población blanca de Norteamérica metaboliza la mefentoína lentamente, incrementando el riesgo del principal efecto adverso, la sedación transitoria. En estas personas, la actividad de otros fármacos biotransformados en mayor o menor grado por la misma enzima que la mefentoína (mefobarbital -anticonvulsivante-, proguanil -antipalúdico- y, posiblemente, diazepam -ansiolítico-) puede verse incrementada.
Casi un 50% de los japonenses, chinos y otros grupos asiáticos carecen de la aldehído deshidrogenasa-2, una enzima implicada en el metabolismo del etanol. En estas personas, la ingestión de etanol ocasiona la acumulación de acetaldehído en plasma, lo que provoca enrojecimiento cutáneo facial, aumento del gasto cardíaco, diaforesis y debilidad muscular. Si las concentraciones de acetaldehído persisten elevadas, puede ocasionar vasodilatación mediada por catecolaminas y síntomas eufóricos y disfóricos.
En alrededor del 85% de los japoneses, chinos y otros grupos de origen asiático; en 5-10% de los ingleses; 9-14% de alemanes y 20% de suizos, la alcohol deshidrogenasa (otra enzima implicada en el metabolismo del etanol) funciona alrededor de 5 veces más rápida de lo normal. Cuando estas personas ingieren alcohol, se acumula acetaldehído, lo que provoca vasodilatación extensa, enrojecimiento facial y taquicardia compensadora.
Déficit de glucosa-6-fosfato deshidrogenasa (G6PD). En los hematíes esta enzima es esencial para que se produzcan las reacciones de reducción indispensables para mantener la integridad de su citoesqueleto. Los pacientes con un déficit de G6PD (aproximadamente el 10% de los varones de raza negra) tienen un riesgo aumentado de padecer una anemia hemolítica cuando se les administran fármacos oxidantes como los antimaláricos cloroquina, pamaquina y primaquina u otros como la aspirina, el probenecid o la vitamina K.
Déficit de glutatión-sintetasa. Esta enzima se encuentra en los hematíes y los hepatocitos. De manera similar al déficit de G6PD (pero ésta ocurre con mucha menor frecuencia), la falta de glutatión-sintetasa en los hematíes ocasiona la aparición de anemia hemolítica tras la administración de fármacos oxidantes. Cuando los hepatocitos tienen una concentración reducida de esta enzima, los pacientes tienen un riesgo superior de lesiones hepáticas tras la administración de fármacos como el paracetamol y la nitrofurantoína.
VARIACIONES FARMACODINÁMICAS
Reducción de la actividad de la warfarina. Algunas personas presentan una respuesta anticoagulante especialmente escasa tras la administración de las dosis terapéuticas habituales de warfarina; para conseguir el efecto farmacológico deseado puede ser necesario administrar una dosis hasta 20 veces superior a la normal. Puesto que en estos casos la biotransformación no es anormal, este defecto genético puede deberse a una reducción de la afinidad de fijación de la warfarina a su receptor.
Hipertermia maligna. Tras recibir una combinación de un relajante muscular depolarizante (por lo general succinilcolina) y un anestésico general potente por vía inhalatoria (halotano por lo general, isoflurano, sevoflurano), algunos enfermos sufren una respuesta hipermetabólica con elevación de la temperatura corporal que puede poner su vida en peligro. La combinación de estos fármacos es la responsable de una reacción similar en pacientes con distrofia muscular y miotonía.
La hipertermia maligna afecta a 1 de cada 20.000 pacientes. Se trata de una alteración que sigue una herencia autosómica dominante. Las mutaciones en el gen que codifica para el receptor de rianodina (canal de calcio) parece ser la causa de la enfermedad. El mecanismo parece relacionado con la potenciación de la actividad del Ca en el retículo sarcoplásmico del músculo esquelético inducida por el halotano. En pacientes susceptibles, el músculo es hiperreactivo al Ca. Como consecuencia de ello, se aceleran las reacciones bioquímicas inducidas por el Ca, lo que produce contracciones musculares extremas y elevación de la tasa metabólica.
El cuadro de hipertermia maligna puede desarrollarse durante la anestesia o en el postoperatorio inmediato. La presentación clínica varía, dependiendo del fármaco utilizado y de la susceptibilidad de cada paciente. A menudo, el primer signo es la rigidez muscular; además (aparte de la hipertermia), aparecen taquicardia, arritmias, acidosis y shock. Posteriormente pueden desarrollarse hipercaliemia, acidosis respiratoria y metabólica, hipocalcemia, elevación de la CK, mioglobinemia y alteraciones de la coagulación (en particular, coagulación intravascular diseminada).
La mortalidad de esta situación en países desarrollados es de alrededor del 7%. El diagnóstico temprano y la instauración rápida del tratamiento son esenciales para reducir la tasa de mortalidad. Se ha desarrollado un test diagnóstico específico (test de la contractura con cafeína-halotano in vitro), pero en la actualidad sólo se dispone en muy pocos centros de Norteamérica. La prevención de nuevos episodios en los miembros de la familia del paciente puede hacerse con biopsias musculares y niveles de colecistoquinina (están elevados). El tratamiento consiste en administrar dantroleno sódico (comenzando con 2,5 mg/kg i.v.), inmediatamente después de que surjan los primeros síntomas. La cirugía y la anestesia deben suspenderse lo antes posible. También debe corregirse la acidosis metabólica y conseguir un enfriamiento superficial y en profundidad y mejorar la ventilación.
INTERACCIONES FARMACOLÓGICAS
Alteración de los efectos de un fármaco por la administración previa o concurrente de otro fármaco (interacción fármaco-fármaco) o por los alimentos (interacción fármaco-alimento,v. cap. 1).
El resultado de una interacción medicamentosa puede ser que el efecto de uno de los fármacos aumente o disminuya. Las interacciones pueden ser deseables, y se consiguen por medio de la combinación de fármacos (p. ej., en el tratamiento de la hipertensión, del asma, de algunas infecciones o de neoplasias), en las que se emplean dos fármacos o más para incrementar los efectos terapéuticos o reducir la toxicidad. Las interacciones indeseables producen reacciones adversas o un fallo terapéutico (v. tablas 301-1 y 301-2).

Puesto que a menudo es difícil predecir la importancia clínica de las interacciones farmacológicas conocidas o esperadas, debe tenerse en cuenta la posibilidad de que aparezcan. Si una interacción parece posible, deberían considerarse las alternativas terapéuticas; sin embargo, no tendría que negarse un tratamiento a un enfermo sólo por la posibilidad de que se produzca una interacción (v. Manejo de las interacciones, más adelante).




INTERACCIONES FARMACODINÁMICAS
Las interacciones farmacodinámicas aparecen cuando un fármaco altera la sensibilidad o la respuesta de los tejidos a otro fármaco. Los fármacos pueden tener efectos opuestos (antagonistas) o efectos aditivos.
Las interacciones de dos fármacos con efectos opuestos deberían ser las detectadas con más facilidad, pero varios factores pueden impedir la identificación precoz de este antagonismo. Por ejemplo, algunos diuréticos tiacídicos suelen elevar la glucemia. Cuando se prescribe un diurético a un paciente diabético que usa insulina o un hipoglucemiante oral, puede contrarrestarse parcialmente el efecto hipoglucemiante del antidiabético, por lo que se requiere un ajuste de las dosis.
Un ejemplo de interacción entre fármacos con efectos farmacológicos similares es el aumento del efecto depresor sobre el SNC que a menudo se produce cuando se toman dos depresores como ansiolíticos, antipsicóticos, antihistamínicos o bebidas alcohólicas. Estas combinaciones aumentan el riesgo de sedación excesiva y fatiga. Los pacientes ancianos son especialmente susceptibles y tienen un riesgo superior de padecer caídas y lesiones (como la fractura de cadera), aunque los estudios epidemiológicos indican que muchas personas toman combinaciones de este tipo sin sufrir problemas mayores.
Con la administración concomitante de dos o más fármacos con actividad anticolinérgica -antipsicóticos (como la clorpromazina), antiparkinsonianos (como el trihexifenidilo) y/o antidepresivos tricíclicos (como la amitriptilina)-, aparecen efectos anticolinérgicos importantes como sequedad de boca y complicaciones dentales asociadas, visión borrosa y, en pacientes expuestos a temperatura y humedad elevadas, hiperpirexia. En algunas personas, especialmente en los ancianos, este efecto aditivo puede desencadenar un delirio de tipo atropínico que podría interpretarse de manera errónea como un empeoramiento de los síntomas psiquiátricos o como una demencia, y podría acelerar la pérdida de memoria y de la capacidad de valerse por sí mismos (v. cap. 304). Puede ser difícil diferenciar los síntomas de la enfermedad a tratar de los efectos derivados del tratamiento, pero es esencial.
Es frecuente que un paciente tome, sin darse cuenta de ello, dos medicamentos que contienen el mismo AINE. Un paciente a quien se ha prescrito ibuprofeno puede comprar una especialidad OTC (over the counter, en castellano «de libre dispensación») con el mismo fármaco sin saber que los dos productos contienen el mismo principio activo. En estos casos, el riesgo de padecer efectos indeseables se incrementa.
Interacciones en el receptor
La enzima monoaminooxidasa (MAO) es esencial en el metabolismo de las catecolaminas, como la noradrenalina. Los inhibidores de la MAO (fenelzina, tranilcipromina) provocan el acúmulo de noradrenalina en las neuronas adrenérgicas. Los fármacos que producen liberación de noradrenalina (p. ej., aminas simpaticomiméticas de acción indirecta) pueden desencadenar (al encontrarse aquélla a concentraciones superiores a las normales) respuestas exageradas, como cefalea intensa, hipertensión (crisis hipertensiva) y arritmias cardíacas. La mayoría de las aminas simpaticomiméticas (como la anfetamina) sólo se dispensan con receta médica, pero otras (fenilpropanolamina) que interaccionan con los inhibidores de la MAO están en muchas especialidades OTC para el resfriado común, procesos alérgicos y preparados dietéticos. Los pacientes que toman inhibidores de la MAO deben evitar estos medicamentos.
Se han descrito reacciones graves (como crisis hipertensivas) en pacientes tratados con inhibidores de la MAO tras la ingestión de comidas y bebidas con un elevado contenido en tiramina (algunos quesos, bebidas alcohólicas, extractos concentrados de levadura, vainas de judías y arenques en conserva). Este efecto es conocido con el nombre de «efecto queso» o reacción queso (v. inhibidores de la MAO en el cap. 189). La tiramina es normalmente metabolizada por la MAO presente en la pared intestinal y en el hígado, y cuando se inhibe la enzima, la tiramina no metabolizada se acumula y libera la noradrenalina de las neuronas adrenérgicas.
El antineoplásico procarbazina y el antiinfeccioso furazolidona (o, quizá, su metabolito) también pueden inhibir la MAO; con estos fármacos hay que tener las mismas precauciones que con los inhibidores de la MAO. Sin embargo, la furazolidona no produce inhibición enzimática hasta 5d después de iniciar el tratamiento, y al cabo de este tiempo, ya suele haberse completado el ciclo terapéutico. La selegilina es un antiparkinsoniano que actúa inhibiendo selectivamente la MAO tipo B. Empleada a dosis terapéuticas (no excediendo los 10 mg/d), la probabilidad de que la selegilina interaccione con otros fármacos y con los alimentos ricos en tiramina es menos probable que la de los antidepresivos inhibidores de la MAO. Incluso puede interactuar con los antidepresivos tricíclicos, con inhibidores selectivos de la recaptación de serotonina (p. ej., fluoxetina) y meperidina y no debería ser utilizado con estos fármacos. Si la dosis de selegilina es >10 mg/d, su selectividad disminuye y el riesgo de interacciones aumenta.
INTERACCIONES FARMACOCINÉTICAS
Las interacciones farmacocinéticas pueden ser complicadas y difíciles de predecir. Las interacciones se deben principalmente a una alteración de la absorción, de la distribución, del metabolismo o de la excreción, que produce modificaciones en la duración y la cantidad de fármaco disponible en el receptor. El tipo de respuesta no se modifica, sino que sólo varía su magnitud y su duración. Las interacciones farmacocinéticas se pueden predecir fundamentalmente a partir del conocimiento de los fármacos por sí solos o se pueden detectar por la monitorización del paciente de los signos clínicos y por los cambios en las concentraciones plasmáticas del fármaco (v. cap. 303).
Alteración de la absorción gastrointestinal
La absorción GI de un fármaco puede estar reducida y su eficacia (actividad terapéutica) comprometida o retrasada, lo cual es indeseable cuando se requiere un efecto rápido para aliviar síntomas agudos como el dolor.
Alteración del pH. Se requiere un medio ácido para disolver de manera adecuada el ketoconazol tras su administración oral y, por tanto, no debería administrarse simultáneamente un antiácido, un anticolinérgico, un antagonista de los receptores H2 o un inhibidor de la bomba (p. ej., omeprazol). En caso de requerir alguno de estos compuestos, se tendrían que administrar por lo menos 2 h después del ketoconazol.
Formación de complejos y adsorción. Las tetraciclinas pueden combinarse con iones metálicos (p. ej., calcio, magnesio, aluminio, hierro) en la luz Gl para formar complejos de baja absorción. Así, ciertos alimentos (p. ej., la leche) o fármacos (p. ej., antiácidos; productos que contengan magnesio, aluminio y sales de calcio; preparados de hierro) son capaces de reducir significativamente la absorción de la tetraciclina. La absorción de doxiciclina y de minociclina está menos afectada por la presencia de leche u otros alimentos, pero está igualmente reducida por antiácidos que contengan aluminio. Los antiácidos incrementan el pH del contenido GI, lo cual posiblemente contribuya a la reducción de la absorción de las tetraciclinas.
Los antiácidos producen también una marcada reducción de la absorción de derivados fluoroquinolónicos (p. ej., ciprofloxacino), probablemente como resultado de la formación de complejos entre los iones metálicos y el fármaco. El intervalo entre la administración de un antiácido y de una fluoroquinolona debería ser lo más largo posible, al menos 2 h, pero preferiblemente un período más prolongado.
Además de unirse y evitar la reabsorción de los ácidos biliares, la colestiramina y el colestipol pueden unirse a otros fármacos que se encuentran en la luz GI, especialmente los de carácter ácido (p. ej., warfarina). El intervalo entre la administración de colestiramina o colestipol y otro fármaco debería ser lo más largo posible (preferiblemente igual o superior a 4 h).
Algunos antidiarreicos (p. ej., aquellos que contienen atapulgito) adsorben otros fármacos, disminuyendo la absorción. Aunque este tipo de interacciones no son bien conocidas, el intervalo entre la toma de estos preparados y la de otro fármaco debería estar tan separada como sea posible.
Alteración de la movilidad. Al aumentar la movilidad GI, fármacos como la metoclopramida, cisaprida o los catárticos pueden acelerar el paso de los fármacos por la luz GI, lo que supone reducir su absorción, particularmente en el caso de los fármacos que requieran un contacto prolongado con la superficie de absorción y los que sólo se absorben en una zona particular de la mucosa GI. Con las formulaciones de recubrimiento entérico o liberación controlada sucede algo similar.
Al disminuir la movilidad GI, los anticolinérgicos pueden reducir la absorción porque retrasan la disolución y enlentecen el vaciado gástrico o aumentar la absorción porque mantienen al fármaco durante más tiempo en contacto con la superficie óptima de absorción.
Efecto de los alimentos. Los alimentos pueden retrasar o reducir la absorción de muchos fármacos. A menudo, los alimentos enlentecen el vaciado gástrico, pero también pueden afectar la absorción porque se unen a los fármacos, reducen su acceso a lugares donde se absorben o alteran su velocidad de disolución o el pH del contenido GI.
La presencia de alimentos en la luz Gl reduce la absorción de muchos antibióticos. A pesar de que hay excepciones (p. ej., la penicilina V, la amoxicilina, la doxiciclina, la minociclina), generalmente se recomienda que los derivados de penicilina y de tetraciclinas y otros antibióticos (p.ej., algunas formulaciones de eritromicina) se administren por lo menos 1 h antes o 2 h después de las comidas para obtener una absorción óptima. También se ha descrito que los alimentos reducen la absorción de alendronato, astemizol, captopril, didanosina y penicilamina, por lo que es importante administrarlos fuera de las comidas. El zumo de naranja, el café y el agua mineral pueden reducir mucho la absorción y eficacia de alendronato, por tanto debe ser administrado con agua al menos 1/2 h antes de la primera comida o del primer fármaco que se administra.
Los alimentos pueden alterar de manera significativa la actividad de la teofilina cuando se administra una formulación de liberación sostenida o retardada, pero no se ve afectada con formulaciones de liberación inmediata. Por ejemplo, cuando se toma una formulación de liberación sostenida <1 h antes de una comida rica en grasa, hay un aumento significativo de la absorción de teofilina y de su pico de concentraciones plasmáticas, en comparación con la administración en ayunas.
Alteración de la distribución
Puede producirse un desplazamiento de la fijación a las proteínas cuando se administran conjuntamente 2 fármacos capaces de fijarse a ellas, en particular cuando pueden fijarse a los mismos lugares de la misma proteína (desplazamiento competitivo). Hay equilibrio entre las fracciones ligada (inactiva) y libre (activa) de los fármacos. Como el fármaco libre se metaboliza y se excreta, el fármaco unido se va liberando gradualmente para mantener el equilibrio y la respuesta farmacológica. El riesgo de interacciones por desplazamiento proteico es significativo, sobre todo en los fármacos que se fijan mucho a ellas (>90%) y cuyo volumen de distribución aparente es pequeño, en particular durante los primeros días de tratamiento conjunto.
El ácido valproico desplaza a la difenilhidantoína de la unión a las proteinas, inhibiendo su metabolismo. En algunos pacientes en tratamiento con los dos fármacos, la concentración libre de difenilhidantoína aumenta significativamente, produciendo más reacciones adversas, incluso cuando la concentración plasmática total de la difenilhidantoína está dentro del rango terapéutico normal. Contrariamente, la difenilhidantoína puede disminuir las concentraciones séricas del ácido valproico. En tratamientos combinados con ambos fármacos se recomienda monitorización con ajustes de dosis cuando sea necesario.
Los fármacos
ácidos generalmente se unen a la albúmina sérica; los fármacos básicos se unen a
la a1-glucoproteína ácida. (v. Uniones en
cap. 298).
Alteración del metabolismo
(V. también tabla 301-1 y Vías metabólicas en cap. 298.)
Estimulación del metabolismo. Un fármaco puede estimular el metabolismo de otro por medio del incremento de la actividad de las enzimas hepáticas involucradas en su metabolismo (inducción enzimática); por ejemplo, el fenobarbital incrementa el metabolismo de la warfarina, disminuyendo su acción anticoagulante. Para compensar este efecto se debe aumentar la dosis de warfarina, pero si el paciente interrumpe el tratamiento con fenobarbital, se debe disminuir la dosis de warfarina para evitar posibles efectos tóxicos. El empleo de sedantes no barbitúricos (p. ej., una benzodiacepina) elimina este riesgo. El fenobarbital también acelera el metabolismo de otros fármacos (p. ej., las hormonas esteroideas). La inducción enzimática también es producida por otros barbitúricos y diversos fármacos como la carbamazepina, la difenilhidantoína, la rifabutina y la rifampicina. La eficacia de algunos fármacos como la clorpromacina, el diazepam, el propoxifeno o la teofilina puede disminuir en grandes fumadores, porque los hidrocarburos policíclicos del humo de los cigarrillos aumentan la actividad enzimática del hígado.
La piridoxina puede acelerar la decarboxilación de la levodopa a dopamina, su metabolito activo, en los tejidos periféricos. A diferencia de la levodopa, la dopamina no puede atravesar la barrera hematoencefálica para producir su efecto antiparkinsoniano. La combinación de carbidopa (un inhibidor de la descarboxilasa) con levodopa previene el efecto de la piridoxina interfiriendo con la acción de la levodopa.
Inhibición del metabolismo. Un fármaco puede inhibir el metabolismo de otro, prolongando e intensificando su actividad. Por ejemplo, el alopurinol reduce la producción de ácido úrico a través de la inhibición de la enzima xantino-oxidasa, que metaboliza fármacos potencialmente tóxicos como mercaptopurina y azatioprina. La inhibición de la xantina-oxidasa puede aumentar considerablemente el efecto de tales fármacos. Por tanto, cuando se administra alopurinol concomitantemente, es recomendable una reducción de 1/3 a 1/4 de la dosis habitual de mercaptopurina o azatioprina.
La cimetidina inhibe las vías metabólicas oxidativas y es probable que aumente el efecto de otros fármacos que se metabolizan por esta vía (p. ej., la carbamazepina, la difenilhidantoína, la teofilina, la warfarina y la mayoría de las benzodiacepinas [incluyendo el diazepam]). La cimetidina no afecta la acción de benzodiacepinas tales como el lorazepam, el oxazepam y el temazepam que se conjugan con glucuronato. La ranitidina tiene menos afinidad por las enzimas hepáticas oxidativas que la cimetidina y, por tanto, las interacciones clínicamente relevantes son más raras. Los estudios realizados con famotidina y nizatidina sugieren que es poco probable que inhiban las vías metabólicas oxidativas y, por tanto, que interaccionen con otros fármacos por este mecanismo.
Concentraciones plasmáticas elevadas de astemizol o cisaprida pueden producir graves reacciones cardiovasculares (p. ej., «torsades de pointes» y otras arritmias ventriculares). Debido a que estos fármacos son metabolizados en gran parte por enzimas citocromo P-450 hepáticas, sus concentraciones plasmáticas pueden incrementarse cuando esas enzimas son inhibidas por fármacos como ciertos antidepresivos (p. ej., nefazodona), claritromicina, eritromicina, itraconazol, ketoconazol y troleandomicina, incrementando por tanto el riesgo de toxicidad. Por consiguiente, el uso concomitante de astemizol o cisaprida con los fármacos anteriormente mencionados y otros fármacos está contraindicado. Debe tenerse precaución cuando se administran conjuntamente astemizol o cisaprida con cualquier fármaco que inhiba enzimas hepáticas. Con los antihistamínicos no sedantes loratadina y fexofenadina no se ha descrito la aparición de reacciones adversas cardiovasculares graves.
El ritonavir, un potente inhibidor de ciertas enzimas citocromo P-450 hepáticas, puede incrementar marcadamente las concentraciones plasmáticas de fármacos que son metabolizados por esta vía (p. ej., antiarrítmicos, astemizol, la mayoría de las benzodiacepinas, cisaprida). Tales fármacos no se deben administrar conjuntamente con ritonavir. El ritonavir también interactúa con otros muchos fármacos, y su uso concomitante debe realizarse con monitorización estricta y ajustes de dosis cuando sea necesario.
Se ha descrito que la eritromicina inhibe el metabolismo hepático de fármacos como la carbamazepina y la teofilina, aumentando así sus efectos. Las fluoroquinolonas ciprofloxacino, enoxacino y grepafloxacino también aumentan la actividad de la teofilina, presumiblemente a través del mismo mecanismo.
El zumo de pomelo inhibe CYP3A4, una enzima citocromo P-450, y de ahí que incremente la biodisponibilidad de ciertos fármacos (p. ej., felodipina), incrementando sus efectos.
Alteración de la excreción urinaria
Alteración del pH urinario: El pH urinario influye sobre la ionización de los ácidos y las bases débiles y, por tanto, afecta su reabsorción y su excreción. Un fármaco no ionizado difunde con mayor facilidad del filtrado glomerular a la sangre. En la orina ácida hay mayor cantidad de ácido no ionizado que en la orina alcalina, en la cual se encuentra, fundamentalmente, en forma de sal. Por tanto, cuando la orina es ácida, más fármaco ácido (p. ej., salicilato) difunde hacia la sangre, lo que aumenta y, quizá, intensifica su actividad. El riesgo de que aparezca una interacción significativa es mayor en pacientes que toman dosis elevadas de salicilatos (p. ej., para el tratamiento de la artritis). Con fármacos básicos (p. ej., la dextroanfetamina) se producen los efectos contrarios. En un estudio se observó que, cuando el pH urinario se mantenía alrededor de 5, en 16 h se excretaba el 54,5% de una dosis de dextroanfetamina, mientras que cuando el pH se elevaba a 8, sólo se excretaba el 2,9%.
Alteración del transporte activo: El probenecid aumenta las concentraciones plasmáticas y prolonga la actividad de los derivados de la penicilina, fundamentalmente por medio del bloqueo de la secreción tubular. Estas combinaciones se han empleado con finalidades terapéuticas.
Cuando se administra quinidina junto con digoxina, se detectan concentraciones plasmáticas de digoxina significativamente superiores que cuando se administra sola. Al parecer, la quinidina reduce el aclaramiento renal de la digoxina, aunque probablemente también participen otros mecanismos no renales.
Se ha descrito que numerosos AINE incrementan la actividad y la toxicidad del metotrexato. Ha habido notificaciones de toxicidad mortal del metotrexato en pacientes que también recibían ketoprofeno. El ketoprofeno puede inhibir la secreción tubular activa renal del metotrexato, pero también es posible que existan otros mecanismos que contribuyan al aumento de las concentraciones plasmáticas de metotrexato. La mayoría de los pacientes que murieron estaban tomando grandes dosis de metotrexato para el tratamiento de neoplasias; sin embargo, también debe tenerse precaución cuando se utilicen dosis más bajas, y en particular en pacientes con artritis reumatoide ya que se vienen utilizando dosis bajas de metotrexato para su tratamiento y que son pacientes que también están tomando AINE.
PRINCIPIOS DE TRATAMIENTO
Los siguientes principios generales son importantes:
• Los fármacos que producen interacciones clínicamente significativas son los que poseen efectos potentes, los que tienen un margen terapéutico estrecho y los que presentan una curva dosis-respuesta con una pendiente pronunciada (p. ej., citotóxicos, hipotensores e hipoglucemiantes; digoxina; warfarina).
• Puede ser difícil distinguir una interacción farmacológica de los factores fisiopatológicos que afectan la respuesta al tratamiento.
• Es posible que las interacciones esperadas no aparezcan; las interacciones dependen de factores individuales como la dosis o el metabolismo de cada paciente.
• Cuando se monitorizan los efectos del fármaco, para que aparezca una interacción suele ser necesario un cambio de dosis o una modificación del tratamiento y no acostumbra a producir efectos adversos relevantes.
• Cualquier desplazamiento de la unión a proteínas altera la relación entre el fármaco total y el fármaco libre y, por tanto, complica la interpretación clínica de la concentración plasmática total. La concentración plasmática total de los fármacos que circulan unidos a las proteínas en gran proporción no tienen el mismo significado en presencia de fármacos que los desplacen, que en su ausencia. Es imprescindible tenerlo en cuenta ya que cada vez se utiliza más la monitorización de los niveles plasmáticos como ayuda en el control terapéutico de los pacientes tratados con diversos fármacos.
La incidencia y las consecuencias clínicas de las interacciones farmacológicas pueden ser minimizadas de diferentes formas. El prescriptor debería conocer la ingesta total de medicamentos por parte del paciente, incluidos los fármacos prescritos por otros médicos y las especialidades de venta libre. Prescribir los fármacos imprescindibles y a las dosis mínimas posibles. Conocer los efectos deseables e indeseables de todos los fármacos empleados, puesto que el espectro de las interacciones farmacológicas suele encontrarse en ambos tipos de efecto. Si es posible, emplear fármacos para los que la dosificación permita un margen de error considerable. Observar y monitorizar los efectos del fármaco en el paciente, en particular tras cualquier alteración del tratamiento; algunas interacciones (p. ej., las metabólicas que dependen de la inducción enzimática) pueden tardar 1 sem o más en aparecer. Considerar las interacciones farmacológicas como causas posibles de cualquier problema inesperado. Si aparecen respuestas clínicas inesperadas, se deberían monitorizar las concentraciones plasmáticas siempre que sea posible; es aconsejable consultar literatura o a especialistas con conocimientos sobre las interacciones, y debería modificarse la dosis del fármaco hasta conseguir el efecto clínico deseado. Si esto falla, se recomienda cambiar el fármaco por otro que no interacciona con los demás que se han prescrito.
PLACEBO
Sustancias inactivas empleadas en los estudios controlados para compararlas con fármacos presuntamente activos, o bien prescritas para aliviar determinados síntomas o para satisfacer las necesidades de tratamiento del paciente.
Un placebo puede ser una maniobra terapéutica (incluyendo técnicas quirúrgicas o psicológicas), pero este capítulo se limita a los fármacos (en todas sus formas, p. ej., oral, parenteral, tópica).
Efectos de los placebos
Algunas personas creen que los placebos son «medicamentos simulados», supuestamente inertes e inofensivos. En la actualidad sabemos que los placebos pueden tener efectos notables, tanto beneficiosos como dañinos. Las publicaciones médicas están repletas de descripciones sobre el poder del placebo para ayudar a los pacientes con ansiedad, tensión psicológica, melancolía, esquizofrenia, cualquier tipo de dolor, cefalea, tos, insomnio, mareo del viajero, bronquitis crónica, resfriado común, artritis, úlcera péptica, hipertensión, náuseas, demencia senil, etc. Pero el placebo no sólo es capaz de ayudar, sino que se ha asociado a efectos indeseables, como náuseas, cefalea, vértigo, somnolencia, insomnio, fatiga, depresión, parálisis, alucinaciones, escozor, vómitos, temblor, taquicardia, diarrea, palidez, erupciones cutáneas, ataxia y edema, por nombrar algunos.
Algunas personas tienen los rasgos típicos de una verdadera adicción a los placebos: tendencia a incrementar la dosis, un deseo compulsivo de tomar la sustancia y un síndrome de abstinencia cuando se interrumpe la administración del placebo.
Mecanismos del efecto placebo
Los efectos subjetivos y objetivos, deseables o indeseables del placebo parecen estar relacionados con 2 componentes del efecto placebo. El primero es la previsión (habitualmente optimista) de los resultados debido a las expectativas asociadas con la medicación; puede llamarse «sugestión», «fe» o «esperanza». El segundo componente, el cambio espontáneo, que a veces es más importante. Si se ha tomado un placebo antes de la mejoría espontánea, ésta puede atribuirse al placebo; del mismo modo, se le incriminará si un paciente presenta cefalea o una erupción cutánea espontánea tras su ingestión.
Reactivos
al placebo son las personas que responden a la administración de los
placebos; sin embargo, puesto que, virtualmente, en determinadas circunstancias
todos somos sugestionables, posiblemente sea más preciso hablar de un espectro
de reactividad al placebo. No se han establecido correlaciones entre
características de la personalidad y respuestas a los placebos. Sin embargo,
personas con personalidades dependientes que tienden a querer complacer a su
médico pueden ser más propensas a comunicar efectos beneficiosos y es más
probable que los pacientes con personalidad histriónica noten algún tipo de
efecto, beneficioso o adverso (v.
cap. 191). Quizá
los factores más importantes son los relacionados con actitudes específicas
hacia la enfermedad, la medicación y el médico. Por ejemplo, cuando un paciente
con un dolor agudo tiene una actitud favorable frente a los medicamentos y un
médico que se preocupa por él y es de su confianza le administra un placebo, la
respuesta puede ser mejor que la de
un paciente con dolor crónico que considera a los fármacos sustancias peligrosas
a quien un médico malhumorado e indeciso le administra un placebo.
Uso en estudios controlados
En estos estudios, los efectos del placebo deben ser restados del efecto global del tratamiento por el fármaco activo. El fármaco en investigación debe actuar significativamente mejor que el placebo para demostrar su eficacia. En algunos estudios, la mejoría que aporta el placebo suele exceder del 50%, por lo que demostrar la eficacia de un principio activo representa un verdadero desafío.
Uso en terapéutica
En cada maniobra terapéutica existe un elemento placebo; por tanto, el efecto atribuido a un fármaco varía en cada paciente y en cada médico, dependiendo de la reactividad al placebo. Es más probable que aparezca un efecto placebo positivo cuando el paciente y el médico creen que proporcionará un efecto beneficioso. Así, una sustancia activa sin efecto farmacológico sobre el proceso a tratar (p.ej., la vitamina B12 para un paciente con artritis) puede aportar una respuesta favorable. Asimismo, un compuesto moderadamente activo (p. ej., un vasodilatador para un paciente con claudicación intermitente) puede aportar un efecto aumentado.
Dar a un paciente un placebo sin su conocimiento es peligroso y puede alterar la relación médico-enfermo. Si el paciente descubriera el engaño, se sentiría traicionado y perdería la confianza en el médico. Cuando en el engaño están implicados otros médicos y enfermeras (en el caso de un centro médico o de un equipo hospitalario), el potencial para modificar de modo adverso las actitudes y el comportamiento por parte de alguno o del resto de los componentes aumenta las probabilidades de que se descubra. El médico puede malinterpretar la respuesta del paciente. Cuando un paciente responde positivamente a un placebo, el médico puede injustificadamente concluir que los síntomas del paciente no son de origen somático o que están exagerados de manera neurótica. Considerando la disponibilidad de multitud de fármacos que, por lo menos, poseen el potencial de aliviar la mayoría de los síntomas que se observan en la práctica y la posibilidad de destruir la relación médico-enfermo con el uso de placebos, está poco indicado el empleo de placebos puros en la práctica clínica.
Por sus costumbres culturales o psicológicas, algunos pacientes parecen beneficiarse de un fármaco que no necesitan o de una forma particular de dosis (p. ej., una inyección cuando la forma oral es suficiente). Los médicos pueden prescribir «tónicos» vitamínicos o inyecciones de vitamina B12 a menudo equivalentes al placebo, aunque a veces se prescriben tabletas de lactosa o «inyecciones estériles».
Los placebos pueden ser utilizados con el consentimiento del paciente para examinar la necesidad de un fármaco potencialmente tóxico. Por ejemplo, no está claro si un paciente con dolor crónico se está beneficiando de un analgésico potencialmente adictivo. Si el paciente consiente, entonces este dilema se puede resolver alternando el fármaco activo y el placebo.
CUMPLIMIENTO DE LA PRESCRIPCIÓN
Grado en el cual el paciente se ajusta al plan terapéutico.
En estudios sobre conducta de los pacientes, sólo la mitad de los pacientes que acuden a la consulta del médico con una prescripción toman la medicación correctamente. La causa más común de abandono del tratamiento es el olvido, que puede ser descrito más apropiadamente como negación de la enfermedad; teniendo que tomar el fármaco como recuerdo constante de la enfermedad. En la tabla 301-3 están recogidas otras causas de abandono del tratamiento.

Los niños generalmente poseen un índice menor que los adultos en el cumplimiento del plan terapéutico. En un estudio de una infección estreptocócica en niños en la que se prescribió 10d de tratamiento con penicilina, un 56% lo interrumpieron al tercer día, un 71% al sexto día y un 82% en el noveno día. Es más difícil el cumplimiento del plan terapéutico en caso de enfermedades crónicas que requieren tratamientos complejos, tratamientos de larga duración (p.ej.,diabetes juvenil, asma). Los padres no entienden las instrucciones de la prescripción y, de acuerdo con los estudios existentes, olvidan la mitad de la información dada por el médico 15 min después (v. Cumplimiento en cap. 258).
Las personas
mayores pueden tomar varios medicamentos; los planes terapéuticos pueden ser
complejos y difíciles de recordar y cumplir, incrementando así la probabilidad
de aparición de interacciones farmacológicas y de efectos adversos
(v.
cap. 304).
Resultados de la falta de cumplimiento
Incluso la pauta terapéutica más completa y bien diseñada falla si el paciente no cumple la prescripción. El resultado más obvio de la falta de cumplimiento de la prescripción es la falta de curación o alivio de la enfermedad. De acuerdo a una estimación realizada en Estados Unidos, la falta de cumplimiento de la prescripción produce 125.000 muertes cada año por enfermedades cardiovasculares. Si los pacientes tomaran sus fármacos directamente, hasta un 23% de las admisiones en clínicas, un 10% de las admisiones hospitalarias, muchas visitas médicas, muchas pruebas diagnósticas y muchos tratamientos innecesarios se podrían evitar.
La falta de cumplimiento de una prescripción puede empeorar la calidad de vida, así como incrementar el coste de los cuidados médicos. Por ejemplo, una dosis olvidada de un fármaco para el tratamiento del glaucoma puede producir daño en el nervio óptico y ceguera; una dosis olvidada de un fármaco para el tratamiento de enfermedades cardíacas puede producir arritmias e insuficiencia cardíaca; el olvido de la toma de una dosis de un antihipertensivo puede producir un accidente cerebrovascular, y un fallo en las dosis prescritas de un antibiótico puede favorecer la reaparición de la infección y la aparición de resistencias bacterianas.
Modos para mejorar el cumplimiento
Los pacientes cumplen mejor con un plan terapéutico si tienen una buena relación con sus médicos, toman parte de las decisiones que se establecen y los médicos muestran interés en que los pacientes cumplan. Una prescripción con instrucciones y explicaciones claras de por qué el tratamiento es necesario y qué se espera lograr de él (p. ej., beneficios, efectos adversos generales) también favorece el cumplimiento de la prescripción por parte del paciente. La verdad en el médico es un punto crucial.
La actitud positiva de los pacientes por preguntar cuestiones y expresar sus dudas puede ayudarles a aceptar la gravedad de sus enfermedades y de manera inteligente valorar las ventajas y desventajas de un plan terapéutico. Hablar sobre los mecanismos inconscientes de la negación de la enfermedad y cómo influye en el olvido de la toma de la medicación, también puede ayudar a mejorar el cumplimiento de la prescripción por parte del paciente. Los pacientes deberían ser informados de comunicar al médico cualquier efecto inesperado o indeseable que aparezca durante el tratamiento antes de que ellos mismos disminuyan o interrumpan el tratamiento. Los pacientes generalmente tienen buenas razones para no seguir con el tratamiento, y su médico, después de una conversación directa con el paciente hablando sobre el problema, puede establecer una dosis correcta.
Los farmacéuticos y el personal de enfermería pueden detectar y ayudar a resolver algunos problemas de cumplimiento. Por ejemplo, el farmacéutico puede notar que el paciente no va a por una nueva tanda de tratamiento o que la prescripción es incorrecta o poco lógica. Repasando las características de la prescripción con el paciente, los farmacéuticos y el personal de enfermería pueden descubrir malentendidos y temores que tienen los pacientes y resolverlos. Una buena comunicación entre todos los profesionales sanitarios es muy importante.
Los grupos de apoyo para pacientes con determinadas enfermedades pueden ser muy útiles para reforzar los planes terapéuticos y resolver problemas.