
303 / MONITORIZACIÓN DEL TRATAMIENTO FARMACOLÓGICO
No siempre es posible disponer de una medida directa del efecto terapéutico deseado. Para muchos fármacos se emplean otras alternativas: determinación de marcadores bioquímicos u, ocasionalmente, signos de toxicidad (p. ej., acúfenos para salicilatos, nistagmus para difenilhidantoína).
La monitorización de la concentración plasmática es un procedimiento alternativo que puede proporcionar una estimación más fácil y rápida de la dosis requerida que la observación aislada de los efectos terapéuticos. La monitorización puede determinar cuándo se alcanza un rango terapéutico ideal y puede ayudar a mantenerlo.

Las concentraciones plasmáticas pueden servir algunas veces como guía primaria en la estrategia de administración de fármacos (v. tabla 303-1). La utilización de la monitorización de las concentraciones plasmáticas puede depender del fármaco (v. tabla 303-2) o de la situación clínica (v. tabla 303-3).

La frecuencia de la monitorización depende del fármaco, de la precisión de medidas previas y de cambios en diferentes factores que afectan la respuesta al fármaco. Por ejemplo, en un paciente con insuficiencia cardíaca, los niveles de digoxina deben ser medidos sólo ocasionalmente si la salud del paciente es estable y si el tratamiento establecido no se modifica; sin embargo, deben ser medidos más frecuentemente si la salud se deteriora o si se añaden fármacos que interaccionan con la digoxina (p. ej., amiodarona, quinidina). Los niveles de teofilina probablemente deberían ser monitorizados a diario en pacientes en servicios de cuidados intensivos, en especial si presentan insuficiencia cardíaca, edema pulmonar u obstrucción aérea grave. En este caso, el metabolismo del fármaco está reducido y varía de manera importante con el tiempo en el mismo paciente.
«VENTANA» TERAPÉUTICA
Intervalo de concentraciones plasmáticas con mayor probabilidad de ejercer un efecto terapéutico.
A pesar de que su aplicación suele ser general, la ventana terapéutica de una población típica a veces resulta inapropiada para un paciente determinado.
En los fármacos que se
fijan a las proteínas plasmáticas y en situaciones en las que es previsible una
alteración en dicha unión, debe ajustarse la concentración total (fijada +
libre) para obtener
la concentración libre deseada. Por ejemplo, las situaciones en las que se halla
reducida la unión a la albúmina (a la que se fijan muchos fármacos ácidos) son:
enfermedad renal terminal, cirrosis, hipoalbuminemia, quemaduras graves y
embarazo, y se ha observado que durante el estrés aumenta la fijación a la
glucoproteína ácida-a1 y a las lipoproteínas (a las que se unen muchos fármacos
alcalinos), mientras que disminuye en las hepatopatías crónicas. En estas
circunstancias, el ajuste de la ventana terapéutica se consigue estimando la
fracción libre en plasma en el paciente y comparándola con la fracción libre en
condiciones normales. Así:

(La fracción libre habitual y la concentración habitual se refieren a la población general, mientras que la fracción libre esperada se refiere al paciente en particular.) Para la difenilhidantoína, la fracción libre habitual puede aumentar de 0,1 a un valor esperado de aproximadamente 0,25 cuando hay una enfermedad renal grave. Por tanto, la ventana terapéutica normal para la concentración total, 7 a 20 mg/l (30 a 80 m mol/l) se debe ajustar a 3 a 8 mg/l (10 a 30 m mol/l) para alcanzar concentraciones libres similares.
EVALUACIÓN DE UNA CONCENTRACIÓN PLASMÁTICA
La evaluación de una concentración medida requiere la obtención de una serie de datos (v. tabla 303-4). La necesidad de informaciones adicionales (p. ej., el estado de las funciones renal, hepática y cardiovascular; la concentración de proteínas séricas, los metabolitos activos y el método analítico) varía según el fármaco a estudiar y cada situación particular.

Interpretación de los datos
Pueden seguirse dos caminos. Uno consiste en comparar el valor observado con el que puede predecirse a partir de la información conocida, para identificar problemas como el incumplimiento de la prescripción, alteraciones de la biodisponibilidad o una eliminación anormalmente lenta o rápida; el otro método consiste en determinar los parámetros farmacocinéticos del fármaco en el paciente, análisis que resulta particularmente útil para determinar la dosis requerida por el paciente.
A partir de la pauta y del momento en que se toma la muestra, se debe establecer inmediatamente si el valor obtenido es un buen estimador de la concentración mínima, media o máxima en equilibrio en una pauta de dosis e intervalos fijos o si es un valor fuera del equilibrio, obtenido poco después de iniciar la administración o tras una pauta irregular.
Equilibrio estacionario. Un valor que represente una estimación de la concentración media en equilibrio tras una pauta a dosis e intervalos fijos resulta particularmente útil. Requiere la obtención de una muestra de plasma por lo menos al cabo de 3 semividas. Además, la fluctuación de la concentración entre las administraciones debe ser pequeña, especialmente si la muestra se extrae justo antes de administrar la dosis siguiente. Esta condición puede cumplirse si el intervalo de administración es <1 semivida (p. ej., la administración diaria de fenobarbital, cuya semivida es 4d). Entonces, la concentración obtenida puede compararse con la esperada.
La concentración media esperada (Cav) está en función de los valores esperados de la biodisponibilidad (F), el aclaramiento (CL) y la velocidad de administración (dosis [D] dividido por el intervalo de la dosis [t], o D/t):
|
|
F
|
|
D
|
|
|
Cav(esperada)=
|
---
|
(esperada) •
|
--
|
(1)
|
|
|
CL
|
|
t
|
|
Si el cociente entre las concentraciones observada y esperada es >1, se absorbe más fármaco del esperado y/o se elimina menos. Cuando el cociente es <1, cualquiera de las situaciones inversas puede ser cierta. En la tabla 303-5 se resumen las causas de absorción y de eliminación alteradas.

Fluctuación del equilibrio. Las pautas de dosificación de muchos fármacos tienen como consecuencia una fluctuación notable en la concentración plasmática. El intervalo de dosificación puede ser comparable o mayor que la semivida plasmática o bien seguir una pauta del tipo 9-1-5-9, en la que el fármaco se toma cada 4 h durante 4 dosis, seguido de un intervalo de 12 h.
En los fármacos cuya pauta implica una fluctuación amplia, el momento preferible para realizar la extracción es justo antes de la dosis siguiente. La concentración mínima (Cmin) esperada al final del intervalo a dosis fijas que sigue a la administración de la dosis (D) en condiciones de equilibrio es:
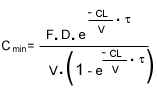 .....(2)
.....(2)
donde V es el volumen de distribución aparente. De nuevo, se puede comparar la concentración observada con la supuesta a partir de los valores esperados de los parámetros.
A menudo, una concentración máxima, a partir de una muestra obtenida justo tras la inyección i.v. o durante el pico de concentraciones tras una dosis oral, es poco fidedigna ya que tanto la absorción como la distribución, o ambas, pueden requerir cierto tiempo para completarse y porque pueden existir variaciones intraindividuales e interindividuales. Sin embargo, cuando la absorción y la distribución son rápidas (p. ej., tras la administración i.m. de aminoglucósidos), medir la concentración plasmática justo después de administrar la dosis y cerca del pico es útil si esta concentración se correlaciona con el efecto del fármaco.
Estado de no-equilibrio. Una muestra sanguínea se puede obtener cuando el fármaco todavía no se ha acumulado o tras unas pautas o unos intervalos de dosificación erráticos. En estas circunstancias no es posible aplicar los principios de la situación de equilibrio; sin embargo, pueden emplearse otros métodos (v. más adelante).
Estimación de los parámetros
Para ajustar la dosis de un paciente, el método más útil consiste en estimar el valor del aclaramiento y, a veces, los valores del volumen de distribución aparente y de la semivida a partir de la monitorización de concentraciones. El aclaramiento es el parámetro más útil porque puede ser utilizado para predecir la relación entre la dosis requerida para alcanzar una concentración plasmática determinada.
A partir de una concentración en equilibrio. Cuando la concentración plasmática es un buen estimador de una concentración media en equilibrio, la biodisponibilidad no varía, se asegura el cumplimiento y el paciente ha seguido una pauta de administración regular y a dosis fijas, el aclaramiento puede obtenerse a partir de la ecuación 1:
|
CL
|
|
D/t
|
|
|
---
|
=
|
---
|
(3)
|
|
F
|
|
Cav
|
Por ejemplo, si se obtiene una concentración de 1,2 mg/l de digoxina (1,5 nmol/l) a partir de la administración crónica de 0,125 mg/d i.v., el aclaramiento/biodisponibilidad es de 104 l/d (4,33 l/h).
Cuando se obtiene una concentración mínima en estado de equilibrio, pueden esperarse fluctuaciones importantes (el intervalo de dosificación es mayor que la semivida) y el fármaco se administra por vía i.v. o su absorción es rápida, el aclaramiento puede calcularse a partir de la relación:
|
|
V
|
|
(
|
F • D |
|
|
)
|
||
|
CL
|
=
|
---
|
• 1n
|
--------
|
+
|
1
|
(4)
|
||
|
|
t
|
|
V • C
|
|
|
(ln = logaritmo natural). Por ejemplo, la gentamicina es un fármaco cuya concentración fluctúa enormemente debido a que el intervalo habitual de dosificación (8 h) es mayor que la semivida habitual (2 h). Si en un paciente de 70 kg se obtiene una concentración mínima de 3 mg/l (6,3 m mol/l) a partir de una pauta i.v. de 80 mg cada 8 h, el aclaramiento sería de 2,0 l/h (F = 1, V = 0,25l/kg). La confianza que merezca esta estimación depende de diversos factores, como la variabilidad del volumen aparente de distribución, de la biodisponibilidad, de los errores de la determinación y los supuestos señalados con anterioridad.
La dosis requerida para alcanzar una concentración media determinada puede calcularse a partir de las estimaciones del aclaramiento y de la biodisponibilidad, según la fórmula siguiente:
|
|
|
CL
|
||
|
D/t
|
=
|
------
|
•Cav |
(5)
|
|
|
|
F
|
En el ejemplo anterior de la digoxina, la dosis diaria v.o. requerida para alcanzar una concentración de 0,6 y 2,0 mg/l -límites de la ventana terapéutica- son 0,062 y 0,21 mg/d (v. ecuación 3). Para la gentamicina, la dosis i.v. cada 12 h (biodisponibilidad = 1) necesaria para mantener una concentración media de 3 mg/l es de 72 mg. Entonces, el mínimo, basado en la ecuación 2, es 1,4mg/l (2,9 m mol/l).
A partir de una concentración obtenida en fase de no-equilibrio. El aclaramiento también se puede estimar a partir de la concentración en fase de no-equilibrio. Por ejemplo, considérense las interpretaciones que se darían a las concentraciones de teofilina obtenidas en 3 pacientes distintos de 70 kg, 12 h después de iniciar una perfusión i.v. de 36 mg/h (45 mg/h de aminofilina). El aclaramiento de la teofilina es muy variable, mientras que su volumen de distribución es relativamente constante (0,5 l/kg). Los pacientes A, B y C tienen aclaramientos de 20, 40 y 80 ml/min, respectivamente. Para que la concentración plasmática del paciente A alcance los niveles tóxicos se requiere una duración de la perfusión >24 h. La concentración en el paciente B se acerca al valor en equilibrio de la ventana terapéutica. Finalmente, en el paciente C, las concentraciones son subterapéuticas (v. fig. 303-1).
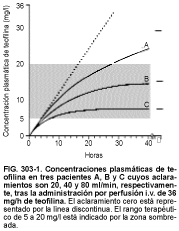
En el caso de una muestra de sangre obtenida 12 h después de iniciar la perfusión, la concentración en el paciente C se acerca al valor en equilibrio y puede estimarse el aclaramiento con bastante precisión empleando las ecuaciones para las situaciones de equilibrio. Las concentraciones de los pacientes A y B no se encuentran próximas al equilibrio, pero los niveles obtenidos ya proporcionan cierta información. La concentración esperada en este momento puede calcularse a partir de:
|
|
R
|
(
|
|
-CL |
|
)
|
||
|
C
|
=
|
----- •
|
1 - e
|
----
|
• tinf
|
(6)
|
||
|
|
CL
|
|
V
|
|
donde R es la velocidad de perfusión y tinf su duración. La concentración al alcanzar el equilibrio es R/CL. La concentración observada en el paciente B es idéntica a la calculada, lo que indica que se acercará a una concentración en equilibrio aproximada de 15 mg/l (83 m mol/l). Sin embargo, en el paciente A, el valor observado de 12 mg/l (67 m mol/l) se encuentra por encima del valor esperado en este momento y cercano a la que se observaría si no hubiese eliminación (15 mg/l), si el aclaramiento es cero (indicada por la línea discontinua en la fig. 303-1). Como consecuencia de los errores analíticos y de otra naturaleza, las únicas conclusiones válidas que se pueden extraer son que el paciente puede aproximarse a los valores potencialmente tóxicos y, por lo tanto, debería considerarse la posibilidad de reducir la dosis y tomar una muestra posterior.
No obstante, la evolución de la concentración estable actual es impredecible.
Cinética dependiente de la concentración
Para fármacos cuyo aclaramiento depende de la concentración (p. ej., difenilhidantoína), no es posible aplicar los principios anteriores, aunque, debido a su comportamiento cinético, la monitorización es particularmente útil. En este caso se aplican los conceptos de la cinética enzimática de Michaelis-Menten.
Factores que complican la monitorización
La presencia
de metabolitos activos complica la monitorización plasmática. Por ejemplo, el
antiarrítmico procainamida es acetilado para formar un metabolito activo, la
N-acetilprocainamida. Los índices de acetilación varían debido a diferencias
genéticas (v. Acetilación en variaciones farmacocinéticas en el
cap. 301). Cerca de las
2/3 partes del fármaco se excretan por orina sin cambios (como procainamida); el
resto es excretado por vía renal en su forma acetilada. De esta manera, la
correlación entre la respuesta terapéutica y la concentración plasmática de la
procainamida en acetiladores rápidos con deterioro de la función renal será
diferente que en aquellos acetiladores lentos con una función renal normal.
Tanto procainamida como su metabolito activo deberían ser monitorizados, en
especial en enfermos renales.
El retraso en la respuesta farmacológica puede también complicar la monitorización. El efecto de la digoxina en el corazón suele estar retrasada ya que el fármaco necesita un tiempo para redistribuirse a sus sitios activos. Por tanto, las concentraciones de digoxina no deberían ser medidas en las primeras 6 h después de su administración, incluso en administraciones por vía i.v. En ese tiempo, las concentraciones plasmáticas no reflejan las concentraciones activas y varían rápidamente.