
12 / METABOLISMO DEL AGUA, LOS ELECTRÓLITOS, LOS MINERALES Y EL EQUILIBRIO ACIDOBÁSICO
El volumen de los líquidos corporales, la concentración de los electrólitos y el equilibrio acidobásico se mantienen normalmente dentro de límites muy estrechos a pesar de las amplias variaciones en la ingesta dietética, la actividad metabólica y las exigencias ambientales. La homeostasis de los líquidos corporales se conserva sobre todo por la acción de los riñones y está controlada por diversos mecanismos fisiológicos interrelacionados. Este capítulo resume muchos aspectos de esos mecanismos en estado de salud, de sus respuestas a las exigencias de la homeostasis y el diagnóstico y tratamiento de varios trastornos comúnmente presentes de los líquidos, los electrólitos y el equilibrio acidobásico.
METABOLISMO DEL AGUA Y DEL SODIO
Agua
El contenido de agua corporal total (ACT) es en promedio un 60% del peso corporal en los hombres jóvenes. El tejido graso tiene un contenido de agua menor; así, la fracción del ACT es en promedio algo inferior en las mujeres (55%) y es considerablemente más baja en las personas obesas y en los ancianos. Alrededor de 2/3 del ACT es intracelular y 1/3 es extracelular. En torno a 3/4 del líquido extracelular (LEC) reside en el espacio intersticial y en los tejidos conjuntivos que rodean las células, mientras que 1/4 es intravascular.
Ingesta: La cantidad de agua ingerida puede variar considerablemente de un día a otro. La ingestión está influida en gran parte por la costumbre, los factores culturales, el acceso a las bebidas y la sed. El margen de volumen de agua que puede ingerirse está determinado por la capacidad del riñón para concentrar y diluir la orina. Un adulto medio con función renal normal necesita de 400 a 500 ml de agua para excretar una carga de solutos diaria en una orina de concentración máxima. Además del agua ingerida, se forman de 200 a 300 ml/d de agua mediante el catabolismo tisular, reduciendo así a un mínimo muy bajo (200 a 300 ml/d) la ingesta de agua imprescindible para evitar la insuficiencia renal. Sin embargo, se necesita una ingesta diaria de agua de 700 a 800 ml para igualar las pérdidas de agua totales y permanecer en equilibrio hídrico (v. más adelante). Una ingesta crónica <700 a 800 ml producirá un aumento de la osmolalidad y un estímulo de la sed. Si la carga de soluto fuera excretada en una orina con dilución máxima se acercaría a un volumen de 25 litros. Una ingesta crónica >25 litros de agua al día llevaría con el tiempo a una pérdida de la homeostasis de los líquidos corporales y a una disminución de la osmolalidad plasmática.
Pérdidas: Las pérdidas insensibles de agua debidas a la evaporación se producen por el aire espirado y la piel, y constituyen entre 0,4 y 0,5 ml/h/kg de peso corporal o unos 650 a 850 ml/24 h en un adulto medio de 70 kg. Cuando hay fiebre pueden perderse de 50 a 75 ml/d adicionales por cada grado centígrado de elevación de la temperatura sobre la normal. Las pérdidas por el sudor son generalmente insignificantes, pero en los climas más cálidos pueden ser importantes si hay fiebre. Las pérdidas de agua por el tracto GI también son despreciables en condiciones de salud, pero pueden ser importantes en las diarreas graves o los vómitos prolongados.
Osmolalidad
Existen importantes diferencias en la composición iónica del líquido intracelular (LIC) y el LEC. El principal catión intracelular es el potasio (K), con una concentración media de 140 mEq/l. La concentración de K extracelular, aunque es muy importante y está estrictamente regulada,
es muy inferior, de unos 3,5 a 5 mEq/l. El principal catión extracelular es el sodio (Na+), con una concentración media de 140 mEq/l. La concentración intracelular de Na+ es mucho más baja, de unos 12 mEq/l. Estas diferencias son mantenidas por la bomba de iones Na+, K+-ATPasa localizada en la práctica totalidad de las membranas celulares. Esta bomba consumidora de energía acopla el movimiento del Na+ hacia fuera de la célula con el movimiento del K hacia el interior de la célula utilizando la energía almacenada en el ATP.
El movimiento del agua entre los compartimientos intracelular y extracelular está controlado en gran parte por la osmolalidad de cada uno de los departamentos, porque la mayoría de las membranas celulares son sumamente permeables al agua. En condiciones normales, la osmolalidad del LEC (290 mOsm/kg de agua) es aproximadamente igual a la del LIC. Por tanto, la osmolalidad plasmática es una guía práctica y precisa de la osmolalidad intracelular. La osmolalidad de los líquidos corporales se puede calcular aproximadamente mediante la siguiente fórmula:
Osmolalidad plasmática (mOsm/kg) =
|
|
|
[Glucosa] |
|
[BUN] |
|
2 [(Na+) sérico] |
+ |
------------ |
+ |
-------- |
|
|
|
18 |
|
2,8 |
donde la (Na+) sérica está expresada en mEq/l y la concentración de glucosa y el BUN en m g/dl. Como señala esta fórmula, la concentración de Na+ es el principal determinante de la osmolalidad plasmática. Por consiguiente, la hipernatremia indica generalmente hipertonicidad plasmática y celular (deshidratación). La hiponatremia suele indicar hipotonicidad plasmática y celular.
Normalmente la osmolalidad plasmática no se ve afectada demasiado por las concentraciones de glucosa o BUN. Sin embargo, la hiperglucemia o una azoemia importante pueden elevar la osmolalidad plasmática en algunas situaciones. En una hiperglucemia intensa, la osmolalidad del LEC aumenta y supera a la del LIC, puesto que la glucosa atraviesa lentamente las membranas celulares en ausencia de insulina, lo que produce un movimiento de agua que sale de las células hacia el LEC. La concentración de Na+ disminuye proporcionalmente a la dilución del LEC, descendiendo 1,6 mEq/l por cada 100 m g/dl (5,55 m mol/l) de incremento en el nivel de glucosa plasmática por encima del normal. Este trastorno se ha denominado hiponatremia por desplazamiento, puesto que no ha tenido lugar ningún cambio neto del agua corporal total (ACT). No está indicado ningún tratamiento específico, porque la concentración de Na+ volverá a ser normal en cuanto la concentración plasmática de glucosa disminuya. A diferencia de la glucosa, la urea penetra con facilidad en las células; dado que la concentración intracelular de urea es igual a la concentración extracelular, no se produce ningún cambio importante del volumen celular. En la azoemia, por consiguiente, aunque aumenta la osmolalidad plasmática, la tonicidad, u osmolalidad plasmática «efectiva», no cambia.
Por último, los cambios que se presentan en la osmolalidad plasmática pueden ser la consecuencia de errores en la determinación del Na+ sérico. Puede existir una seudohiponatremia plasmática con una osmolalidad plasmática normal en la hiperlipidemia o en casos de hiperproteinemia extrema, puesto que los lípidos o las proteínas ocupan parte del espacio en el volumen de la muestra obtenida para el análisis. Los métodos más recientes de determinación de los electrólitos plasmáticos con electrodos iónicos selectivos soslayan este problema.
La osmolalidad plasmática se puede determinar directamente. Existe un hiato osmolar cuando la osmolalidad plasmática medida supera la calculada mediante la fórmula anterior en >10 mOsm/l. La presencia de un aumento del hiato osmolar puede deberse a una o más sustancias osmóticamente activas medidas en el plasma. La tabla 12-1 recoge algunas de las causas más frecuentes de un aumento del hiato osmolar. Cuando se encuentra este aumento deben realizarse inmediatamente más pruebas de laboratorio específicas para determinar la causa e iniciar el tratamiento específico.
El volumen de ACT está regulado por la sed, la secreción de hormona antidiurética (ADH) y los riñones. Los osmorreceptores situados en el hipotálamo anterolateral son estimulados por la elevación de la osmolalidad plasmática y estimulan a los centros de la sed adyacentes. El estímulo de la sed produce la percepción consciente de la sed y aumenta en consecuencia la ingesta de agua. Los osmorreceptores responden asimismo a la hiperosmolalidad mediante la inducción de la liberación de ADH por la hipófisis posterior. La secreción de ADH causa a su vez un aumento de la reabsorción de agua en la nefrona distal al aumentar la permeabilidad de este segmento de la nefrona, por lo demás relativamente impermeable al agua. La osmolalidad del LEC se mantiene normalmente dentro de unos límites estrechos. Un aumento de un 2% produce sed y liberación de ADH. Además del aumento de la osmolalidad plasmática, puede tener lugar una estimulación no osmótica de la liberación de ADH. En casos de grave depleción del volumen, se secreta ADH para preservar el LEC con independencia de la osmolalidad plasmática. En esta situación, el agua se retiene a expensas de la osmolalidad plasmática.
Sodio
Dado que el sodio (Na+) es el principial catión osmóticamente activo en el compartimiento del LEC, los cambios en el contenido total de Na+ del organismo van seguidos de cambios correspondientes en el volumen del LEC. Cuando el contenido total de Na+ es bajo, el volumen del LEC resulta deplecionado. Esta depleción del volumen del LEC es detectada por los barorreceptores situados en las aurículas del corazón y las venas torácicas y causa un aumento de la retención renal de Na+. Cuando el contenido total de Na+ es alto, aparece una sobrecarga de la volemia. Los receptores de presiones altas localizados en el seno carotídeo y el aparato yuxtaglomerular detectan la sobrecarga y aumentan la natriuresis para que la volemia pueda ajustarse a la normalidad.
El contenido total de Na+ del organismo está regulado por un equilibrio entre la ingesta dietética y la excreción renal del Na+. Una depleción de Na+ importante no se produce a no ser que existan pérdidas de Na anormales renales o extrarrenales -por la piel o el tracto GI-, combinadas con una ingesta insuficiente de Na+. Los defectos de la conservación renal del Na+ también pueden originarse por una nefropatía primaria, por insuficiencia suprarrenal o por un tratamiento diurético. Análogamente se produce una sobrecarga de Na+ si hay un desequilibrio entre la ingesta y la excreción, pero dada la gran capacidad excretora de Na+ de los riñones normales, una sobrecarga de Na+ implica por lo general una excreción renal de Na+ defectuosa.
La excreción renal de Na+ puede ajustarse con amplitud de acuerdo con la ingesta de Na+. El control de la excreción renal de Na+ se inicia con el ajuste del flujo sanguíneo renal y la TFG. La cantidad de Na+ transportada a la nefrona para su reabsorción es directamente proporcional a la TFG. Por consiguiente, puede haber una retención de Na+ secundaria a la insuficiencia renal. Igualmente, la disminución del flujo sanguíneo renal, como en la insuficiencia cardíaca, reducirá la TFG y la carga de Na+ filtrada, con producción de edemas.
El eje renina-angiotensina-aldosterona es probablemente el principal mecanismo regulador de la excreción de sal. En los estados de depleción de la volemia, la TFG y el aporte de Na+ a la nefrona distal disminuyen, causando liberación de renina por las células arteriolares aferentes del aparato yuxtaglomerular. El angiotensinógeno (sustrato de la renina) es escindido enzimáticamente por la renina para formar el polipéptido inactivo angiotensina I. La angiotensina I es escindida una vez más por la enzima conversora de angiotensina (ECA) a la hormona activa angiotensina II. Ésta aumenta la reabsorción de Na+ mediante una disminución de la carga de Na+ filtrada y potenciando la reabsorción de Na+ en el túbulo proximal. La angiotensi-na II estimula también a las células de la corteza suprarrenal a secretar el mineralocorticoide aldosterona. La aldosterona eleva la reabsorción de Na+ por medio de efectos directos sobre el asa de Henle, el túbulo distal y el túbulo colector. Las alteraciones de la regulación del eje renina-angiotensina-aldosterona conducen a diversos trastornos del volumen de líquido y los electrólitos. La actuación farmacológica sobre el sistema renina-angiotensina-aldosterona sigue siendo el pilar principal del tratamiento de muchos de esos trastornos.
Recientemente se han identificado varios factores natriuréticos, entre ellos una sustancia análoga a la ouabaína que induce la natriuresis por inhibición de la Na+, K+-ATPasa. También se ha identificado un segundo grupo de péptidos natriuréticos auriculares (PNA). Los PNA circulantes activos parecen contener 28 aminoácidos y se derivan del extremo C-terminal de un péptido precursor. Los PNA se encuentran en los gránulos secretores del tejido auricular del corazón y parecen ser liberados en respuesta a aumentos agudos de la presión inducidos por la PA, a la carga de sal y a la expansión del volumen del LEC y a otras causas de distensión auricular. Se han descrito niveles elevados de PNA en el plasma en Pacientes con sobrecarga de volumen del LEC, aldosteronismo primario, insuficiencia cardíaca, insuficiencia renal, cirrosis con ascitis y en algunos Pacientes con hipertensión esencial.
A la inversa, se ha observado disminución de los niveles plasmáticos de PNA en algunos Pacientes con síndrome nefrótico y un supuesto aumento del volumen efectivo del LEC circulante.
Los PNA antagonizan in vitro los efectos vasoconstrictores de la angiotensina II e inhiben la acción conservadora del Na+ de la aldosterona. Cuando se infunden PNA en animales o en seres humanos, los efectos son variables. La infusión de niveles fisiológicos de PNA induce una leve natriuresis en seres humanos, pero también reduce los niveles plasmáticos de la angiotensina II, la aldosterona y la actividad de la renina plasmática. Dosis mayores de PNA aumentan la natriuresis y elevan la TFG a pesar de la caída del flujo plasmático renal y de la PA. Los PNA parecen representar un papel importante en la regulación del volumen del LEC, el metabolismo del Na+ y la PA. Sin embargo, sigue sin aclararse su plena significación fisiológica, fisiopatológica y terapéutica.
TRASTORNOS DEL METABOLISMO DEL AGUA Y DEL SODIO
Aunque los trastornos del equilibrio del agua y del Na+ suelen presentarse juntos, es útil considerarlos por separado.
Equilibrio del agua: El agua corporal total (ACT) está distribuida entre el LIC (2/3) y el LEC (1/3). Los déficits o los excesos puros de agua se distribuyen entre el LIC y el LEC aproximadamente en la misma proporción. En consecuencia, los signos clínicos de alteración del volumen del LEC no suelen ser llamativos en los trastornos puros del ACT; en lugar de ello,
los signos están relacionados por lo general con los cambios de la osmolalidad del LEC. Dado que la concentración de Na+ sérico es el principal determinante de la osmolalidad del LEC, en la hiponatremia se produce hiperhidratación, mientras que en la hipernatremia se produce deshidratación. El término deshidratación se suele usar para referirse a un déficit combinado de Na+ y de ACT, pero es una mejor descripción de la depleción relativamente pura de ACT. Hiperhidratación es la mejor descripción de un aumento relativamente puro del ACT.
Equilibrio del sodio: Dado que el Na+ está restringido sobre todo al LEC, los déficits o los excesos del contenido total de Na+ del organismo se caracterizan respectivamente por signos de depleción o de sobrecarga del volumen del LEC. La concentración sérica de Na+ no se modifica necesariamente con los déficits o los excesos del Na+ corporal total.
La determinación del estado del volumen del LEC se apoya exclusivamente en la exploración física. La presión venosa central (PVC) se puede calcular sumando 5 mm Hg a la altura de la pulsación venosa de la yugular interna por encima del segundo espacio intercostal con la cabeza y el tronco del Paciente elevados 30º mientras descansa en decúbito supino. La PVC puede medirse directamente utilizando un catéter venoso central situado en la aurícula derecha o la vena cava superior. La PVC normal es de 1 a 8 cm de H2O (1 a 6 mm Hg). Esta determinación indica de forma fiable el estado del volumen intravascular, salvo si el Paciente tiene taponamiento pericárdico, disfunción de la válvula tricúspide, insuficiencia ventricular izquierda aguda o insuficiencia cardíaca derecha pura. Si estos trastornos están presentes, la presión capilar pulmonar de enclavamiento valora con más precisión la presión de llenado del ventrículo izquierdo y el volumen intravascular efectivo. La presión capilar pulmonar de enclavamiento es normalmente de 6 a 13 cm H2O (5 a 10 mm Hg). Además de un aumento de la PVC, el exceso de volumen del LEC también causa edema. En un adulto medio de 70 kg tiene que acumularse un aumento del LEC de unos 3 litros antes de que el edema pueda detectarse en la exploración física. Si se descartan las causas locales de edema, como obstrucción venosa o linfática, la presencia de edema es un signo fiable de exceso de Na+. Las manifestaciones adicionales de exceso de Na+, como el edema pulmonar, dependen en gran parte del estado cardíaco y de la distribución del LEC entre los espacios vascular e intersticial.
Contracción del volumen de líquido extracelular
Disminución del volumen del LEC causada por una disminución neta del contenido total de sodio corporal.
Patogenia
Las pérdidas de Na+ del organismo están siempre combinadas con pérdidas de agua. Por consiguiente, el resultado final de la depleción de Na+ es una depleción del volumen del LEC. Que la concentración de Na+ plasmático aumente, disminuya o permanezca constante con depleción del volumen depende en gran parte de la vía de la pérdida de volumen (p. ej., GI, renal) y del tipo de líquido de reposición ingerido por la persona o que se le administra. Otros factores que pueden afectar también a la concentración plasmática de Na+ en una depleción de volumen son, entre otros, la secreción de ADH o la disminución del aporte de soluto al túbulo distal, cuyo resultado es la retención de agua. En la tabla 12-2 se enumeran las causas frecuentes de una depleción del volumen del LEC.

Síntomas, signos y diagnóstico
La depleción de volumen del LEC debe sospecharse en los Pacientes con historia de ingesta líquida insuficiente (especialmente en Pacientes comatosos o desorientados), vómitos, diarrea (o pérdidas GI yatrogénicas, p. ej., aspiración nasogástrica, ileostomía o colostomía), tratamiento diurético, síntomas de diabetes mellitus y enfermedad renal o suprarrenal. A veces se obtiene también una historia reciente de pérdida de peso.
En una depleción leve del volumen del LEC, los únicos signos pueden ser la disminución de turgencia de la piel y de la tensión intraocular. La sequedad de las mucosas no suele ser fiable, especialmente en los ancianos y en los Pacientes que respiran por la boca. Son signos más fiables la hipotensión ortostática (disminución de la presión sistólica en >10 mm Hg al ponerse de pie) y la taquicardia y una PVC baja, aunque la hipotensión ortostática puede presentarse en Pacientes encamados sin depleción de volumen del LEC. Cuando el volumen del LEC ha disminuido aproximadamente en un 5% o más, la taquicardia y/o la hipotensión ortostáticas están generalmente presentes. Una depleción grave del volumen del LEC puede producir desorientación y un shock manifiesto.
Unos riñones funcionalmente normales responden a la depleción del volumen del LEC con una retención de Na+. Cuando la depleción del volumen es lo bastante grave como para producir una disminución del volumen de orina, la concentración de Na+ urinario suele ser <10 a 15 mEq/l; la fracción de excreción de Na+ (Na+ urinario/Na+ sérico dividido por creatinina urinaria/creatinina sérica) suele ser <1%, y la osmolalidad de la orina suele estar elevada. Si además de la depleción del volumen del LEC existe alcalosis metabólica, la concentración urinaria de Na+ puede ser alta y, por ello, engañosa como medida del estado del volumen; en este caso, una concentración baja del cloruro (Cl) urinario (<10 mEq/l) indica de modo más fiable una depleción del volumen del LEC. Si las pérdidas de Na+ son debidas a nefropatía, diuréticos o insuficiencia suprarrenal, la concentración urinaria de Na+ es generalmente >20 mEq/l. Datos de laboratorio como el hematócrito aumentan a menudo en la depleción de volumen, pero son difíciles de interpretar a no ser que se conozcan los valores basales. Una depleción importante del volumen de LEC produce con frecuencia aumentos leves o moderados en los niveles plasmáticos de BUN y creatinina (azoemia prerrenal; cociente BUN/creatinina >20:1).
Tratamiento
La depleción del volumen del LEC leve o moderada puede corregirse aumentando la ingesta oral de Na+ y agua si el Paciente está consciente y no tiene disfunción GI. Hay que corregir la causa subyacente a la depleción de volumen mediante la interrupción de los diuréticos o tratando la diarrea. Cuando la depleción de volumen es grave y se asocia con hipotensión, o cuando no es posible la administración oral de líquidos, la primera opción es el suero salino i.v. (v. más adelante las precauciones descritas). Cuando la excreción renal de agua es normal, el déficit de Na+ y agua puede reemplazarse sin peligro con solución salina al 0,9%. Cuando existe un trastorno asociado en el metabolismo del agua, los líquidos de reposición se modifican en la forma que se expone en las secciones siguientes. Cuando la depleción del volumen del LEC se debe a trastornos metabólicos como la cetoacidosis diabética o la enfermedad de Addison, o está complicada por ellas, debe prestarse atención a la corrección de estos problemas además de a la reposición del volumen. (V. también caps. 9 y 13.)
Expansión del volumen de líquido extracelular
Aumento del volumen del LEC causado por un aumento neto del contenido total de sodio del organismo asociado con formación de edema.
Patogenia
Dado que el Na+ está en gran medida limitado al LEC, los aumentos del contenido de Na+ total del organismo se reflejan en incrementos subsiguientes del volumen del LEC. Los aumentos del volumen intravascular suelen conducir a incrementos inmediatos en la excreción renal de Na+ y agua. Por consiguiente, el mantenimiento de una sobrecarga del volumen del LEC y la formación de edema implican un secuestro de líquido en el espacio intersticial. El movimiento de los líquidos entre los espacios intersticial e intravascular es una función de las fuerzas de Starling en los capilares. Un aumento de la presión hidrostática capilar, como ocurre en la insuficiencia cardíaca; una disminución de la presión oncótica plasmática, como ocurre en el síndrome nefrótico, o una combinación de ambos, como ocurre en la cirrosis hepática grave, producen un movimiento neto de líquido hacia el espacio intersticial y la formación de edema. En esas enfermedades, la depleción de volumen subsiguiente conduce a un aumento de la retención de Na+ por los riñones y al mantenimiento de un estado de sobrecarga. Las causas frecuentes de sobrecarga de volumen se enumeran en la tabla 12-3.

Síntomas, signos y diagnóstico
Los síntomas iniciales de sobrecarga de volumen del LEC son bastante inespecíficos y pueden presentarse antes de una formación manifiesta de edema. Entre ellos están la ganancia de peso y la debilidad. Cuando la sobrecarga de volumen es causada por insuficiencia del ventrículo izquierdo, pueden presentarse también tempranamente disnea de esfuerzo, reducción de la tolerancia al ejercicio, ortopnea y disnea paroxística nocturna.
En los comienzos de la formación de edema son frecuentes síntomas como los ojos hinchados al levantarse por la mañana y los zapatos apretados al final del día. En la insuficiencia cardíaca los edemas suelen presentarse en partes declives y pueden acompañarse de una multitud de hallazgos físicos, como estertores pulmonares, PVC elevada, ritmo de galope en S3 y corazón agrandado con edema pulmonar y/o derrames pleurales en la radiografía del tórax. El edema se limita con frecuencia a las extremidades inferiores y se asocia con ascitis en la cirrosis hepática. Suelen existir signos asociados con la cirrosis, como angiomas vasculares, ginecomastia, eritema palmar y atrofia testicular. En contraste, el edema suele ser difuso en el síndrome nefrótico y a veces va acompañado de anasarca generalizado con derrames pleurales y ascitis. El edema periorbitario se observa con frecuencia, pero no de forma constante, en el síndrome nefrótico.
Tratamiento
El tratamiento inicial debe orientarse a la corrección de la causa subyacente a la expansión de volumen del LEC. Es preciso tratar la disfunción ventricular izquierda, la isquema miocárdica y las arritmias cardíacas. El uso de la digital, los agentes inotrópicos y la reducción de la poscarga para mejorar la función cardíaca pueden disminuir o evitar la necesidad de diuréticos a través del aumento del aporte de sodio a los riñones y reduciendo de ese modo la retención de Na+ renal. El tratamiento de las causas subyacentes al síndrome nefrótico es variable y depende de la histopatología renal específica presente. Varias de las causas subyacentes al síndrome nefrótico no responden al tratamiento; no obstante, a menudo puede reducirse el nivel de proteinuria mediante un inhibidor de la ECA. Los inhibidores de la ECA tienen que emplearse siempre con precaución en Pacientes con deterioro renal que necesitan ser monitorizados por empeoramiento de la insuficiencia renal e hiperpotasemia.
Los diuréticos son de gran utilidad en la sobrecarga de volumen del LEC. Los diuréticos del asa, como la furosemida, inhiben la reabsorción de Na+ en la rama ascendente gruesa del asa de Henle. Los diuréticos tiazídicos inhiben la reabsorción de Na+ en el túbulo distal. Tanto los diuréticos del asa como las tiazidas conducen a una pérdida de K, que puede ser problemática en algunos Pacientes. Los diuréticos ahorradores de K, como la amilorida, el triamtereno y la espironolactona, inhiben la reabsorción de Na+ en la parte distal de la nefrona y en el túbulo colector. Cuando se usan solos, tienen unos efectos natriuréticos moderados. El triamtereno y la amilorida se han utilizado conjuntamente con una tiazida para prevenir la pérdida de K.
La resistencia a los diuréticos es un hecho corriente; el origen es con frecuencia multifactorial. En la mayoría de los casos, las principales causas que contribuyen a la resistencia son el tratamiento insuficiente de la etiología subyacente a la sobrecarga del volumen, el incumplimiento por parte del Paciente (especialmente respecto a la restricción dietética de sal), la depleción de volumen del LEC y la insuficiencia renal. Las dosis escalonadas de diuréticos del asa suelen tener éxito en la promoción de la diuresis. También ha resultado útil combinar un diurético del asa y una tiazida para tratar a los Pacientes resistentes a la diuresis con uno u otra por separado.
Una vez corregida la sobrecarga de volumen mediante la natriuresis, el mantenimiento de la euvolemia requiere restricción del Na+ dietético (principalmente por medio del ajuste de los hábitos dietéticos), a no ser que pueda eliminarse totalmente la enfermedad subyacente. Suele ser difícil determinar la ingesta de Na+ por medio de la historia clínica, pero puede monitorizarse con éxito midiendo el Na+ urinario en una muestra de orina de 24 h una vez alcanzado el estado estacionario (es decir, en ausencia de cambios de peso o cambios recientes de la dosis de diuréticos). Las dietas con un contenido de 2 a 3 g/d de Na+ se toleran muy bien y actúan razonablemente en todos los casos de sobrecarga de volumen, salvo en los muy graves. Generalmente se utilizan sales de potasio como sustitutos de la sal para ayudar a los Pacientes a tolerar la dieta baja en sal; sin embargo, se debe tener precaución, especialmente en el caso de los Pacientes que reciben diuréticos ahorrado-res de K o inhibidores de la ECA y en los que tienen insuficiencia renal, porque puede producirse una hiperpotasemia potencialmente mortal.
Hiponatremia
Disminución de la concentración de sodio plasmático por debajo de 136 mEq/l causada por un exceso de agua en proporción a los solutos.
Incidencia, etiología y patogenia
La hiponatremia es el más frecuente de los trastornos electrolíticos, con una incidencia de hasta un 1% de los Pacientes que ingresan en el hospital. La hiponatremia se ha descrito en más de un 50% de los Pacientes hospitalizados con SIDA.
La hiponatremia refleja un exceso de ACT en relación con el contenido total de Na+ corporal. Dado que este contenido se refleja en el estado del volumen del LEC, es útil clasificar las causas de hiponatremia con las de hipovolemia, volumen del LEC normal e hipervolemia. En la tabla 12-4 se resumen las principales causas de hiponatremia.

Las pérdidas de líquido renales que llevan a una hiponatremia pueden producirse en la deficiencia de mineralocorticoides, en el tratamiento diurético, la diuresis osmótica o la nefropatía con pérdida de sal. Esta clase de nefropatía abarca un grupo de nefropatías intrínsecas, vagamente definido, con disfunción principalmente intersticial (tubular). Tales enfermedades son la nefritis intersticial, la enfermedad quística medular, la obstrucción parcial del tracto urinario y, a veces, la nefropatía poliquística. Generalmente las causas renales de hiponatremia hipovolémica se pueden diferenciar de las causas extrarrenales por medio de la historia clínica. Los Pacientes con pérdidas renales continuas de líquidos se diferencian de aquellos con pérdidas de líquido extrarrenales porque tienen una concentración de Na+ urinario inadecuadamente alta (>20 mEq/l). Una excepción a esto se presenta en la alcalosis metabólica (como ocurre en los vómitos prolongados), donde se pierden con la orina grandes cantidades de HCO3 que fuerzan a una excreción de Na+ para mantener la electroneutralidad. En la alcalosis metabólica, la concentración de Cl urinario diferencia muchas veces el origen renal o extrarrenal de la depleción de volumen (v. Alcalosis metabólica, más adelante).
Los diuréticos pueden producir también hiponatremia hipovolémica. Los diuréticos tiazídicos, en particular, afectan a la capacidad de dilución de los riñones a la vez que aumentan la excreción de Na+. Una vez que se presenta la depleción de volumen, la liberación no osmótica de ADH puede causar retención de agua y empeorar la hiponatremia. La hipopotasemia concomitante desplaza Na+ hacia el interior de la célula y potencia la liberación de ADH, agravando con ello la hiponatremia. Este efecto de las tiazidas puede durar hasta 2 sem tras la interrupción del tratamiento; por otra parte, la hiponatremia suele responder a la reposición del K y el déficit de volumen junto con una prudente restricción de la ingesta de agua hasta que desaparezca el efecto del fármaco. Los Pacientes ancianos pueden ser especialmente susceptibles a la hiponatremia inducida por las tiazidas, especialmente si existe un defecto preexistente de la excreción renal de agua. En raras ocasiones, esos Pacientes pueden desarrollar una hiponatremia grave, peligrosa para la vida, pocas semanas después del inicio del uso del diurético tiazídico, como consecuencia de una natriuresis exagerada y de un deterioro subyacente de la capacidad de dilución de la orina. Los diuréticos del asa causan hiponatremia con una frecuencia mucho menor.
Existen muchas causas posibles de hiponatremia en el SIDA debido a los múltiples sistemas orgánicos afectados en esa enfermedad. La hiponatremia puede originarse por la administración de líquidos hipotónicos en caso de deterioro de la función renal o bien por la liberación no osmótica de vasopresina debida a depleción del volumen intravascular, con o sin administración coincidente de fármacos que reducen la excreción renal de agua. Además, la insuficiencia suprarrenal se ha hecho cada vez más frecuente en los Pacientes con SIDA como consecuencia de adrenalitis por citomegalovirus, infección por micobacterias o interferencia con la síntesis de glucocorticoides y mineralocorticoides renales debida a ketoconazol. Por último, el síndrome de secreción inapropiada de ADH (SIADH) puede presentarse en Pacientes con SIDA debido a infecciones coexistentes pulmonares o del SNC.
La hiponatremia hipovolémica se caracteriza por deficiencias de agua y de Na+, aunque proporcionalmente se haya perdido más Na+ que agua. La hiponatremia puede presentarse cuando las pérdidas de líquido como las que se producen por vómitos prolongados, diarrea grave o secuestro de líquidos en el tercer espacio son repuestas con la ingestión de agua libre o tratadas con líquido i.v. hipotónico. También se producen pérdidas importantes del LEC con la liberación no osmótica de ADH, que causa retención de agua por los riñones y el mantenimiento de la hiponatremia o un nuevo empeoramiento de la misma. La hiponatremia hipovolémica puede producirse por pérdidas de líquido tanto extrarrenales como renales. Las pérdidas extrarrenales son las GI y las del tercer espacio. Pueden secuestrarse volúmenes de líquido sorprendentemente grandes en la pancreatitis, la peritonitis y la obstrucción del intestino delgado, o pérdidas en las quemaduras graves de grandes áreas de la superficie corporal. La respuesta renal normal a la pérdida de volumen es la conservación del Na+. En las causas de hipovolemia extrarrenales se produce típicamente una concentración urinaria de Na+ <10 mEq/l.
La hiponatremia euvolémica se produce cuando aumenta el ACT y no existe un cambio importante del contenido total de Na+ corporal. La polidipsia primaria sólo puede causar hiponatremia si la ingesta de agua desborda la capacidad de los riñones para excretarla. Dado que los riñones normales pueden excretar hasta 25 litros de orina/d, la hiponatremia debida exclusivamente a la polidipsia se produce sólo por la ingestión de grandes cantidades de agua o por defectos de la capacidad de dilución renal. Esto suele ocurrir sólo en casos de psicosis o en Pacientes con grados más moderados de polidipsia e insuficiencia renal. La hiponatremia dilucional puede ser también el resultado de una ingesta de agua excesiva sin retención de Na+ en presencia de insuficiencia renal, enfermedad de Addison, mixedema o secreción no osmótica de ADH (p. ej., por estrés, estados postoperatorios y fármacos como clorpropamida o tolbutamida, opiáceos, barbitúricos, vincristina, clofibrato y carbamazepina). La hiponatremia postoperatoria ocurre hasta en un 4,5% de los Pacientes como consecuencia de una combinación de liberación de ADH no osmótica y administración excesiva de líquidos hipotónicos después de la cirugía. Ciertos fármacos, como la ciclofosfamida, los AINE y la clorpropamida, potencian el efecto renal de la ADH endógena, mientras que otros, como la oxitocina, tienen un efecto directo análogo al de la ADH sobre el riñón. Una deficiencia de excreción de agua es común a todos esos trastornos.
La hiponatremia hipervolémica se caracteriza por un aumento del contenido total de Na+ corporal y también del ACT. Diversos trastornos edematosos, como la insuficiencia cardíaca y la cirrosis hepática, se asocian con hiponatremia hipervolémica. La hiponatremia ocurre raras veces en el síndrome nefrótico, pero debe tenerse presente la seudohiponatremia debida a la interferencia en la determinación del Na+ por los lípidos elevados. En cada uno de estos trastornos, la disminución del volumen circulante efectivo conduce a la liberación de ADH y angiotensina II. La hiponatremia, si existe, es el resultado del efecto antidiurético de la ADH sobre el riñón, así como del deterioro de la excreción renal de agua por la angiotensina. La disminución de la TFG y la estimulación de la sed potencian también el desarrollo de hiponatremia. Además de la hiponatremia y el edema, las concentraciones de Na+ bajas (<10 mEq/l) y la alta osmolalidad urinaria (en relación con el plasma) se producen generalmente en ausencia de diuréticos.
Efectos sobre el SNC: Experimentalmente, el contenido de agua celular del cerebro está elevado tanto en la hiponatremia aguda como en la crónica; sin embargo, como consecuencia de la disminución del contenido de electrólitos de la célula cerebral, el aumento del contenido de agua del cerebro es menor del que sería de esperar para el grado de osmolalidad plasmática. En la hiponatremia aguda, las células cerebrales no pueden ajustar su tonicidad con normalidad y se produce hinchazón. Por ello, los síntomas de disfunción del SNC son más frecuentes y la mortalidad es considerablemente mayor en la hiponatremia aguda que en la crónica.
El síndrome de secreción inadecuada de ADH (SIADH) está definido por una orina diluida en un grado inferior en presencia de hipoosmolalidad e hiponatremia plasmáticas. Además, el diagnóstico se apoya en la ausencia de depleción o sobrecarga de volumen, estrés emocional o dolor y diuréticos u otros fármacos que estimulan la secreción de ADH y en la presencia de función cardíaca, hepática, renal, suprarrenal y tiroidea normales. La SIADH se asocia con multitud de trastornos (v. tabla 12-5).

Aunque el SIADH se atribuye clásicamente a una liberación sostenida de ADH, se han identificado varias pautas anormales de liberación de ADH mediante radioinmunoensayo. En algunos Pacientes, la liberación de ADH es irregular y aparentemente independiente del control osmótico. En otro gran subgrupo, los niveles de ADH varían adecuadamente con la osmolalidad plasmática, pero el umbral osmótico para la liberación de ADH es anormalmente bajo (un reajuste del osmostato). Un pequeño subgrupo de Pacientes parecen tener un grado de liberación de ADH constante a un nivel bajo; dentro del margen normal de la osmolalidad plasmática, la liberación de ADH es adecuada, pero cuando el plasma se hace hipoosmótico la liberación de ADH no se inhibe. Otro pequeño subgrupo de Pacientes no pueden alcanzar la dilución máxima de la orina o excretar una carga de agua, pero tienen una liberación de ADH normal. Estos Pacientes tienen un síndrome de antidiuresis inadecuada en lugar de un SIADH y sólo pueden identificarse mediante la determinación de los niveles de ADH en plasma.
Síntomas y signos
Los síntomas de hiponatremia se presentan generalmente cuando la osmolalidad plasmática efectiva desciende a £240 mOsm/kg con independencia de la causa subyacente. La tasa de descenso, sin embargo, puede ser tan importante como la magnitud absoluta del mismo; los síntomas pueden aparecer con osmolalidades plasmáticas algo más elevadas si el cambio tiene lugar con rapidez. Las manifestaciones de hiponatremia pueden ser poco perceptibles y consisten sobre todo en cambios de la situación mental, como alteración de la personalidad, letargo y confusión. Cuando la hiponatremia va acompañada de alteraciones en el contenido total de Na+ corporal, también existen signos de depleción o sobrecarga de volumen (v. más atrás Contracción del volumen de líquido extracelular y Expansión del volumen de líquido extracelular). Cuando el Na+ plasmático cae por debajo de 115 mEq/l, puede producirse estupor, hiperexcitabilidad neuromuscular, convulsiones, coma prolongado y muerte. Raras veces, la mejoría inicial en respuesta al tratamiento puede ir seguida de síntomas neurológicos tardíos que culminan en coma, estado vegetativo persistente o muerte. Se han observado diversos cambios anatómicos, como edema cerebral, herniación de la amígdala cerebelosa y lesiones desmielinizantes (tanto pontinas como extrapontinas). Se han descrito cambios neuropatológicos de mielinólisis pontina central asociados con la hiponatremia, especialmente en Pacientes con alcoholismo, malnutrición u otras afecciones debilitantes crónicas. Se discute la relación de la mielinólisis con la rapidez y el grado de corrección de la hiponatremia o con la anoxia (v. Tratamiento, más adelante).
Pruebas recientes indican que las mujeres premenopáusicas que tienen ciclos menstruales pueden ser especialmente susceptibles a un edema cerebral grave en asociación con hiponatremia aguda, tal vez debido a inhibición de la Na+, K+-ATPasa cerebral por los estrógenos y la progesterona que produce la disminución de la salida de solutos de las células cerebrales. Las secuelas descritas son el infarto hipotalámico y de la hipófisis posterior y la herniación del tallo encefálico en los casos graves.
Pronóstico
La mortalidad es considerablemente mayor en la hiponatremia aguda que en la crónica debido a los efectos sobre el SNC, como se comentó antes. La presencia de afecciones debilitantes también parece tener una influencia sobre la supervivencia en la hiponatremia. Así, la mortalidad aumenta cuando la hiponatremia está asociada con alcoholismo, cirrosis hepática o procesos malignos.
Tratamiento
El tratamiento de la hiponatremia leve y asintomática (es decir, Na+ plasmático >120 mEq/l) es sencillo, en especial si es posible identificar la causa subyacente y suprimirla. Así, en Pacientes con hiponatremia inducida por tiazidas, la supresión del diurético y la reposición de las deficiencias de Na+ y/o K puede bastar. Análogamente, si una hiponatremia leve es consecuencia de la administración inadecuada de líquidos por vía parenteral en un Paciente con dificultad para la excreción de agua, puede ser suficiente con interrumpir simplemente el tratamiento con líquidos hipotónicos.
La presencia de hiponatremia, hiperpotasemia e hipotensión debe sugerir una insuficiencia suprarrenal y puede requerir la administración i.v. de glucocorticoides (100 a 200 mg de hidrocortisona en 1 litro de solución de dextrosa al 5% en solución salina al 0,9% a lo largo de 4 h para el tratamiento de la insuficiencia suprarrenal aguda, v. también Tratamiento en Enfermedad de Addison, cap. 9). Cuando la función suprarrenal es normal, pero la hiponatremia está asociada con depleción de volumen del LEC e hipotensión, la administración de solución salina al 0,9% suele corregir la hiponatremia y también la hipotensión. Si el trastorno subyacente responde con lentitud o la hiponatremia es intensa (es decir, Na+ plasmático <120 mEq/l), es muy eficaz la restricción de la ingestión de agua a no más de 500 a 1.000 ml/24 h.
La mayoría de los Pacientes en los que la hiponatremia dilucional se asocia con una expansión de volumen del LEC debida a la retención renal de Na+ (p. ej., insuficiencia cardíaca, cirrosis o síndrome nefrótico) tienen pocos síntomas relacionados con la hiponatremia. En estos casos suele tener éxito la restricción de agua combinada con el tratamiento de la enfermedad subyacente. En Pacientes con insuficiencia cardíaca, el captopril, un inhibidor de la ECA, junto con un diurético del asa, puede corregir la hiponatremia refractaria al tratamiento. El captopril y otros inhibidores de la ECA también pueden ser eficaces en estados de expansión del volumen del LEC caracterizados por aumento de actividad del eje renina-angiotensina-aldosterona, en especial el síndrome nefrótico. Si está presente un SIADH, es necesaria una intensa restricción, de un 25 a un 50%, de agua de mantenimiento. La duración de la restricción depende del éxito del tratamiento del proceso patológico subyacente.
Cuando la enfermedad subyacente deja de ser susceptible de tratamiento, como en el caso de un cáncer pulmonar metastásico, y la intensa restricción de agua es inaceptable para el Paciente, puede ser útil la demeclociclina (900 a 1.200 m g/d); sin embargo, la demeclociclina se ha asociado con insuficiencia renal aguda en Pacientes con cirrosis hepática. Aunque la disfunción renal suele ser reversible tras la supresión de la demeclociclina, el fármaco debe evitarse en Pacientes con cirrosis y utilizarse con precaución en otras situaciones.
El tratamiento de la hiponatremia es más discutido cuando se presentan síntomas de intoxicación acuosa grave (es decir, convulsiones) o la hiponatremia es intensa (Na+ plasmático <115 mEq/l; osmolalidad efectiva <230 mOsm/l). La discusión se refiere principalmente al ritmo y al grado de corrección de la hiponatremia. Cuando la hiponatremia es intensa pero asintomática, la restricción rigurosa de la ingesta de agua suele ser inofensiva y suficiente, aunque algunos expertos recomiendan la administración de solución salina hipertónica. Se dispone de solución salina hipertónica (3%) con un contenido de 513 mEq Na+/l para el tratamiento de la hiponatremia grave con síntomas (como en el caso de las convulsiones generalizadas). Debido a la posibilidad de desencadenar secuelas neurológicas adicionales (en particular la mielinólisis pontina central, v. más adelante), la solución salina hipertónica debe utilizarse con gran precaución en situación de hiponatremia. Los especialistas coinciden en que la hipercorrección de la hiponatremia es peligrosa; debe evitarse la hipernatremia e incluso la normonatremia.
Muchos especialistas recomiendan que el Na+ plasmático no se incremente con una rapidez superior a 1 mEq/l/h y que el aumento absoluto no sea mayor de 10 mEq/l/24 h. Lo que sigue es un término medio razonable: se infunden lentamente por vía i.v. 250 ml de solución salina hipertónica (3%) y se determina el sodio sérico 10 h después. Si los valores siguen siendo demasiado bajos la cantidad puede repetirse, manteniendo el aumento del Na+ sérico dentro de los 10 mEq/l/24 h.
En los Pacientes con una expansión coincidente del volumen del LEC (incluidos los que tienen un SIADH), la administración de un diurético del asa como la furosemida puede combinarse con solución salina isotónica más KCl para reponer las pérdidas de K inducidas por el diurético. En la hiponatremia hipervolémica que no responde a los diuréticos puede ser imprescindible la hemofiltración intermitente o continua para controlar el volumen del LEC mientras se corrige la hiponatremia con solución salina hipertónica i.v.
La secuela neurológica más importante consecutiva a una corrección demasiado rápida de la hiponatremia es la mielinólisis pontina central (desmielinización de la protuberancia basal central). La desmielinización puede afectar también a otras áreas del encéfalo. En pocas semanas o meses pueden aparecer una tetraparesia, así como debilidad de la parte inferior de la cara y la lengua. La lesión puede extenderse en dirección dorsal y afectar a los tractos sensitivos y dejar al Paciente con un síndrome de bloqueo (un estado consciente y sensible en el que el Paciente, debido a una parálisis motora generalizada, no puede comunicarse, excepto tal vez mediante movimientos oculares convenidos). El daño suele ser permanente, y pueden sobrevenir complicaciones sistémicas. Si el Na+ se repone con demasiada rapidez (p. ej., cuando se administran grandes volúmenes de solución salina fisiológica a un Paciente quemado), la inducción de hiponatremia con líquido hipotónico a veces puede mitigar el desarrollo de la mielinólisis pontina central.
Hipernatremia
Elevación de la concentración plasmática de sodio por encima de 145 mEq/l causada por un déficit de agua en proporción al soluto.
La hipernatremia de los recién nacidos se expone en Problemas metabólicos del recién nacido, capítulo 260.
Incidencia, patogenia y etiología
La hipernatremia es menos frecuente que la hiponatremia, y se presenta en <1% de los Pacientes ingresados en un hospital de cuidados intensivos; sin embargo, la hipernatremia en un Paciente adulto está entre los trastornos electrolíticos más graves, con una mortalidad descrita del 40 al 60%. Dado que el Na+ es el principal determinante de la osmolalidad del LEC, la hipernatremia implica hiperosmolalidad del compartimiento del LEC. La hipertonicidad del LEC en relación con el LIC determina un movimiento del agua, que sale del espacio intracelular hasta que la tonicidad celular aumenta hasta la del LEC. Las elevaciones del BUN también conducen a hiperosmolalidad del LEC, pero puesto que el BUN atraviesa libremente las membranas celulares, esto no conlleva un movimiento de salida de agua del compartimiento del LIC o los síntomas asociados. Generalmente se produce hipernatremia cuando se pierde agua corporal y no se repone suficientemente. Las pérdidas de agua pueden ser aisladas o pueden tener lugar en conjunción con pérdidas de Na+. Por tanto, la hipernatremia puede estar asociada con depleción de volumen del LEC, con euvolemia o con sobrecarga de volumen del LEC. Con independencia del estado del volumen, la hipernatremia implica habitualmente o bien un deterioro del mecanismo de la sed o bien un acceso limitado al agua. La gravedad de los procesos patológicos subyacentes que suelen conducir a una incapacidad para beber se considera responsable en parte de la alta mortalidad que se observa en la hipernatremia. En la tabla 12-6 se enumeran las causas más frecuentes de hipernatremia.

La hipernatremia asociada con una depleción del volumen aparece en las pérdidas de Na+ acompañadas de una pérdida relativamente mayor de agua corporal. Las causas extrarrenales comunes son la mayoría de las que originan hiponatremia y depleción de volumen del LEC (v. más atrás). Puede presentarse hipernatremia o hiponatremia en una pérdida de volumen intensa según las cantidades relativas de sal y de agua perdidas y la cantidad de agua bebida antes de la presentación.
Las causas renales de hipernatremia y depleción de volumen incluyen el tratamiento con diuréticos del asa. Los diuréticos del asa inhiben la reabsorción de Na+ en la porción concentradora de la nefrona y pueden aumentar el aclaramiento de agua. La diuresis osmótica puede conducir también a un deterioro de la capacidad concentradora renal debido a la presencia de una sustancia hipertónica en la luz del túbulo distal. El glicerol, el manitol, y a veces la urea, pueden causar diuresis osmótica con hipernatremia resultante. La causa más frecuente de la hipernatremia debida a diuresis osmótica es posiblemente la hiperglucemia presente junto al coma hiperglucémico-hiperosmolar en los Pacientes diabéticos. Dado que la glucosa no penetra en las células en ausencia de insulina, la hiperglucemia deshidrata aún más el LIC. El grado de hiperosmolalidad puede resultar amortiguado por la reducción artificial del Na+ plasmático resultante del movimiento del agua que sale de las células hacia el LEC (v. más atrás, Osmolalidad). Una enfermedad renal intrínseca como la insuficiencia renal crónica también puede impedir la formación de una orina de concentración máxima y predispone a la hipernatremia.
Cuando existe un déficit exclusivo de agua, la hipernatremia se produce sin alteraciones del equilibrio del Na+. Las causas extrarrenales de pérdidas acuosas, como la sudación excesiva, conducen a alguna pérdida de Na+, pero dado que el sudor es hipotónico, se puede producir hipernatremia antes que una hipovolemia importante. Un déficit puro de agua ocurre también en la diabetes insípida central o nefrógena.
La diabetes insípida hipofisaria o central es un defecto de la producción o la liberación de ADH por la hipófisis posterior. Al igual que en la diabetes insípida, los Pacientes con diabetes insípida central no pueden excretar normalmente una orina concentrada. Dado que el mecanismo de la sed está intacto, en la diabetes insípida central aparece una hipernatremia grave sólo cuando el acceso al agua llega a estar restringido. El defecto de la concentración en la diabetes insípida central es normalmente sensible a la administración exógena de ADH (p. ej., vasopresina, 1-desamino-8-d-arginina-vasopresina y otros, v. Trastornos del lóbulo posterior, cap. 7).
La diabetes insípida nefrógena causa un defecto en la capacidad concentradora de la orina a pesar de la presencia de niveles suficientes del ADH circulante. Como ocurre en la diabetes insípida central, los Pacientes con diabetes insípida nefrógena no pueden concentrar la orina adecuadamente. La sed evita una hipernatremia grave en los Pacientes que tienen acceso al agua, pero a diferencia de la diabetes insípida central no existe respuesta a la ADH exógena (v. Diabetes insípida nefrógena, cap. 229).
La hipernatremia esencial o hipodipsia primaria se presenta a veces en niños con lesión cerebral y en ancianos gravemente enfermos. Se caracteriza por un deterioro del mecanismo de la sed y un desencadenamiento osmótico de la liberación de ADH. La liberación no osmótica de ADH parece mantenerse intacta y estos Pacientes están generalmente euvolémicos.
La hipernatremia se asocia también con sobrecarga de volumen. Generalmente, la hipernatremia se produce por una ingesta de Na+ exageradamente elevada asociada con un acceso limitado al agua. Un ejemplo es la administración excesiva de Na+HCO3 hipertónico durante la RCP o en la acidosis láctica. La hipernatremia también puede producirse por la administración de suero salino hipertónico o por hiperalimentación.
La hipernatremia es especialmente frecuente en los ancianos. Los motivos pueden ser la dificultad de obtener agua, la alteración del mecanismo de la sed, el deterioro de la capacidad de concentración renal (debido a los diuréticos o a la pérdida de nefronas que acompaña al envejecimiento o por otras nefropatías) y al aumento de las pérdidas insensibles. La liberación de ADH está potenciada en respuesta a los estímulos osmóticos, pero disminuye debido a los cambios de volumen y presión en la ancianidad. Además, algunos Pacientes ancianos tienen una alteración de la producción de angiotensina II, lo cual puede contribuir directamente a deteriorar el mecanismo de la sed, la liberación de ADH y la capacidad de concentración del riñón. En los ancianos es especialmente frecuente la hipernatremia, tanto en el período postoperatorio como en los Pacientes que reciben alimentación por sonda, nutrición parenteral u otras soluciones hipertónicas.
Síntomas y signos
El síntoma principal de la hipernatremia es la sed. La falta de sed en los Pacientes conscientes con hipernatremia indica un deterioro del mecanismo de la sed. Los Pacientes con dificultades para comunicarse pueden ser incaPaces de manifestar que tienen sed y obtener con ello acceso al agua. Los principales signos de hipernatremia son consecuencia de disfunción del SNC debida a contracción de volumen de las células cerebrales. Se puede producir confusión, excitabilidad neuromuscular, crisis convulsivas o coma; el daño cerebrovascular con hemorragia subcortical o subaracnoidea y las trombosis venosas son hallazgos frecuentes en la autopsia de Pacientes fallecidos por hipernatremia grave.
Experimentalmente, las sustancias activas osmóticamente en el líquido intracelular del SNC se elevan en respuesta a la hipernatremia crónica. Por tanto, el grado de deshidratación de las células cerebrales y los síntomas resultantes del SNC son menos graves en la hipernatremia crónica que en la aguda.
Cuando la hipernatremia se produce en asociación con alteraciones del equilibrio del Na+, se presentan los síntomas típicos de depleción o sobrecarga del volumen (v. más atrás, Contracción de volumen del líquido extracelular y Expansión de volumen del líquido extracelular). En los Pacientes con defectos de concentración renal se excreta característicamente un gran volumen de orina hipotónica. Cuando las pérdidas son extrarrenales, la vía de la pérdida de agua suele ser manifiesta (p. ej., vómitos, diarrea, sudación excesiva) y la concentración urinaria de Na+ es baja.
Diagnóstico
Una prueba de restricción de agua puede servir para diferenciar varios estados poliúricos. Puesto que esta prueba puede originar una hiperosmolalidad peligrosa, sólo debe llevarse a cabo con una monitorización estricta de las concentraciones de los electrólitos, la osmolalidad sanguínea y urinaria y el estado de la volemia. La ingesta de agua se limita hasta que el Paciente pierde de un 3 a un 5% del peso corporal o hasta que tres determinaciones consecutivas de la osmolalidad urinaria con intervalos de una hora están separadas un 10% entre sí. Se debe tener cuidado para evitar una deshidratación excesiva. Una vez alcanzada la osmolalidad urinaria máxima se inyectan por vía s.c. 5 U de vasopresina acuosa y al cabo de 1 h se determina la osmolalidad urinaria. La respuesta normal a la restricción de agua es una elevación de la osmolalidad urinaria a >800 mOsm/l con poco o ningún aumento ulterior por la acción de la vasopresina.
En una diabetes insípida central completa, la osmolalidad urinaria máxima con restricción de agua es <300 mOsm/l; esta osmolalidad aumenta considerablemente tras la administración de vasopresina. En la diabetes insípida central parcial, la osmolalidad urinaria máxima oscila entre 300 y 800 mOsm/l tras la restricción de agua; se calcula en >10% el aumento de la osmolalidad urinaria previsible con la vasopresina. En la diabetes insípida nefrógena, la osmolalidad urinaria máxima con la restricción de agua es de 300 a 500 mOsm/l; la osmolalidad cambia muy poco en respuesta a la vasopresina. Además, en la diabetes insípida nefrógena los niveles plasmáticos de ADH obtenidos tras la restricción de agua son bastante altos (>5 pg/ml). Los Pacientes con polidipsia primaria tienen respuestas similares a la restricción de agua y a la administración de vasopresina debido a la atenuación del gradiente de concentración de la médula renal. Estos Pacientes pueden diferenciarse de los que tienen diabetes insípida nefrógena por la presencia de niveles plasmáticos de ADH inhibidos que se obtiene tras la restricción de agua (<5 pg/ml).
Pronóstico
La mortalidad de la hipernatremia aguda es considerablemente mayor que la de la hipernatremia crónica. La mortalidad de la hipernatremia, en general, sigue siendo alta debido a los efectos de la hiperosmolalidad en el SNC y a la gravedad de la enfermedad subyacente necesaria que cause una imposibilidad para responder a la satisfacción de la sed.
Tratamiento
La reposición del agua es el principal objetivo del tratamiento. El agua es eficaz administrada por boca en los Pacientes que están conscientes y en ausencia de una disfunción GI de importancia. En la hipernatremia grave o en los Pacientes que no pueden beber por los vómitos continuos o por alteraciones del estado mental se prefiere la hidratación por vía i.v. Aunque a la mayoría de los Pacientes se les puede administrar solución de dextrosa al 5%, una infusión demasiado rápida puede producir glucosuria, aumentando con ello la excreción de agua libre de sales, y una elevación de la hipertonicidad. Si las alteraciones de la volemia son lo bastante graves como para producir shock, puede ser necesario administrar coloides y solución salina al 0,9% para aumentar el volumen del LEC antes de que se administre solución salina hipotónica y agua libre para corregir la hipernatremia. Si la duración de la hipernatremia es <24 h, debe corregirse en 24 h. Sin embargo, si la hipernatremia es crónica o de duración desconocida, debe corregirse a lo largo de más de 48 h y la osmolalidad se debe reducir a un ritmo no mayor de 2 mOsm/l/h para evitar el edema cerebral causado por exceso de solutos en el cerebro. La cantidad de agua necesaria para reponer el déficit existente se puede calcular mediante la fórmula siguiente:
Déficit de agua libre = ACT x [(Na+ plasmático/140) - 1]
donde el ACT se expresa en litros y se calcula multiplicando el peso en kg por 0,6; el Na+ plasmático se expresa en mEq/l. Esta fórmula asume un contenido total de Na+ corporal constante. En los Pacientes con depleción del contenido total de Na+ corporal (es decir, en la depleción de volumen), el déficit de agua libre es superior al calculado mediante la fórmula.
En Pacientes con hiponatremia y sobrecarga de volumen (exceso de contenido total del Na+ corporal), se puede administrar un diurético del asa y reemplazar las pérdidas urinarias sumadas al déficit de agua existente con solución de dextrosa al 5%. Debe administrarse KCl en función de la concentración plasmática de K.
En la diabetes insípida central, la administración de ADH corrige la pérdida renal de agua. El tipo y la vía de administración de la ADH dependen de la gravedad de la hipernatremia y del estado clínico del Paciente. Cuando la hipernatremia es moderada y el Paciente está consciente, se puede administrar desmopresina (1-desamino-8-d-arginina-vasopresina sintética), de 10 a 20 ml cada 12 a 24 h por vía intranasal. La vasopresina acuosa se usa con menor frecuencia, dada la preocuPación sobre sus potenciales efectos secundarios -como el vasospasmo coronario- mediados a través del receptor V1. Por otra parte, la corta duración de la acción de la vasopresina acuosa por vía subcutánea puede ser ventajosa y permite una reducción más controlada del Na+ plasmático en los Pacientes gravemente afectados, especialmente cuando el déficit de agua es grande. El tratamiento prolongado de la diabetes insípida central con vasopresina y otros fármacos (p. ej., clorpropamida) se expone en el capítulo 7.
En la diabetes insípida nefrógena adquirida, el mejor enfoque de tratamiento consiste en suprimir la causa subyacente. Deben corregirse la hipopotasemia y la hipercalcemia; debe suprimirse el litio, la demeclociclina y otras causas posibles de ausencia de respuesta a la ADH. Algunas veces, la supresión del litio no elimina con rapidez la diabetes insípida nefrógena. Se ha descrito que la amilorida reduce en parte la poliuria latente debida a la diabetes insípida nefrógena inducida por el litio. La administración de un diurético tiazídico también es útil junto con una restricción moderada de Na+. Además de los diuréticos pueden ser útiles los AINE, especialmente en las variedades congénitas de la diabetes insípida nefrógena.
En los Pacientes con hipernatremia e hipovolemia, especialmente en los diabéticos con coma hiperglucémico no cetósico, se puede administrar solución salina al 0,45% para reponer el Na+ y el agua libre. Si existe una acidosis grave (pH <7,20), el suero salino al 0,45% puede sustituirse con una solución hipotónica de Na+HCO3. Los Pacientes con una diabetes hiperosmolar suelen responder a pequeñas dosis de insulina. Debe administrarse insulina regular por vía i.v. o i.m. hasta que la glucemia disminuya a 250 m g/dl (13,88 m mol/l) en las primeras horas de tratamiento. El nivel de la glucemia debe monitorizarse con frecuencia durante el tratamiento para evitar una caída demasiado rápida de la glucemia o una hipoglucemia. Las altas dosis de insulina necesarias para tratar la cetoacidosis diabética no suelen ser necesarias en Pacientes con un coma hiperglucémico-hiperosmolar no cetósico y pueden ser realmente perjudiciales, puesto que se asocian con un descenso demasiado rápido de la glucosa plasmática y edema cerebral.
METABOLISMO DEL POTASIO
El potasio (K) es el catión intracelular más abundante. Sólo alrededor de un 2% del K corporal total es extracelular. Dado que la mayoría del K intracelular está en el interior de las células musculares, el K total corporal es aproximadamente proporcional a la masa corporal magra. Un adulto medio de 70 kg tiene en torno a 3.500 mEq de K.
El K es el principal determinante de la osmolalidad intracelular. La proporción entre las concentraciones de K del líquido intracelular y el extracelular influye fuertemente sobre la polarización de la membrana celular, la cual a su vez repercute sobre importantes procesos celulares, como la conducción de los impulsos nerviosos y la contracción de la célula muscular (incluida la miocárdica). Así, alteraciones relativamente pequeñas en la concentración de K plasmático pueden tener manifestaciones clínicas importantes.
En ausencia de alteraciones metabólicas graves, el nivel plasmático de K proporciona una valoración clínica razonable del contenido total de K corporal. Suponiendo constante el pH plasmático, una disminución de la concentración plasmática de K desde 4 a 3 mEq/l indica un déficit de K total de 100 a 200 mEq. Un descenso de K plasmático <3 mEq/l indica un déficit total de unos 200 a 400 mEq. En muchos estados patológicos, la concentración plasmática de K se convierte en una guía poco fiable del contenido total de K corporal porque los procesos involucrados producen desplazamientos de K hacia dentro y hacia fuera de las células.
Equilibrio interno del potasio
Son numerosos los factores que afectan al movimiento del K entre los compartimientos del líquido intracelular y extracelular. Entre los más importantes está el nivel de insulina circulante. El K se desplaza al interior de las células en presencia de insulina, reduciendo de ese modo la concentración plasmática de K. Cuando falta la insulina circulante, como en la cetoacidosis diabética, el K sale de las células, elevando así el K plasmático, incluso cuando existe una deficiencia de K corporal total. La estimulación del sistema nervioso simpático afecta también al movimiento transcelular del K. Los agonistas b-adrenérgicos, en especial los agonistas selectivos b2, promueven la captación celular de K, mientras que un bloqueo b-adrenérgico o un estímulo de los agonistas a parecen estimular el movimiento de K al exterior de las células. El K plasmático puede afectar también de manera importante al pH plasmático. La acidosis metabólica aguda facilita el movimiento del K hacia fuera de las células y al LEC. La alcalosis metabólica aguda estimula la transferencia de K en la dirección opuesta. Sin embargo, los cambios de la concentración plasmática de HCO3 pueden ser más importantes que los cambios del pH a este respecto. Así, la acidosis causada por la acumulación de ácidos minerales (hiato no aniónico, acidosis hiperclorémica) es más probable que muestre una elevación del K plasmático debida a desplazamientos transcelulares. Al contrario, la acidosis metabólica por acumulación de ácidos orgánicos (aumento de acidosis por el hiato aniónico) no causa hiperpotasemia. Por ello, la hiperpotasemia que suele acompañar a la cetoacidosis diabética es resultado de la deficiencia de insulina y de la hipertonicidad del LEC, más que de la acidosis por sí misma. La acidosis y la alcalosis respiratoria agudas parecen tener menos efecto sobre la concentración plasmática de potasio que las alteraciones metabólicas. En cualquier caso, la concentración plasmática de K debe interpretarse siempre en el contexto del pH plasmático (y de la concentración de HCO3).
Equilibrio externo de potasio
La ingesta dietética de K varía normalmente entre 40 y 150 mEq/d. En estado estacionario las pérdidas fecales son relativamente constantes y pequeñas (en torno al 10% de la ingesta). La excreción urinaria está regulada para aproximarse a la ingesta de K, de modo que el equilibrio se mantenga. Sin embargo, cuando la carga de K se ingiere de forma rápida, aparece en la orina sólo un 50% de la carga a lo largo de las horas siguientes. La elevación del K plasmático se reduce al mínimo mediante la transferencia de la mayor parte de la carga de K restante hacia el compartimiento intracelular. Si la ingesta elevada continúa, la excreción renal aumenta debido probablemente a la secreción de aldosterona estimulada por el K. Además, la absorción de K a partir de las heces parece estar bajo cierto grado de regulación y puede disminuir un 50% en caso de exceso de K crónico.
Cuando la ingesta dietética de K disminuye, el K intracelular sirve de nuevo como amortiguador frente a las oscilaciones amplias de la concentración plasmática de K. La conservación renal del K se desarrolla en forma relativamente lenta en respuesta a las disminuciones del K de la dieta, y es mucho menos eficiente que la capacidad de los riñones para conservar el Na+. Una excreción urinaria de K de 10 mEq/24 h representa una capacidad de conservación renal de K próxima a la máxima y, por consiguiente, implica una importante depleción de K.
El K plasmático se filtra libremente en el glomérulo. La mayor parte del K filtrado es reabsorbido en el túbulo proximal y el asa de Henle. El K es secretado hacia el filtrado en el túbulo distal y el túbulo colector. La excreción renal neta de K está regulada principalmente por los cambios en la secreción de K en el segmento distal de la nefrona. La secreción distal de K es regulada por la aldosterona, el estado acidobásico y la tasa de flujo urinario en la nefrona distal, y por la polaridad de la membrana. Los niveles altos de aldosterona circulante conducen a un aumento de la secreción de K y a caliuresis. La deficiencia o la inhibición de la aldosterona reduce la secreción de K en la nefrona distal y causa la conservación del K. La acidosis aguda dificulta la excreción de K, mientras que la acidosis crónica y la alcalosis aguda conducen a caliuresis (v. Alteraciones del metabolismo acidobásico, más adelante). El aumento del aporte de Na+ a la nefrona distal y las altas tasas de flujo urinario en la nefrona distal favorecen la secreción de K. La reabsorción de Na+ en la nefrona distal aumenta la negatividad eléctrica luminal, un factor que favorece aún más la secreción de K. Por tanto, el aumento de oferta de Na+ a la nefrona distal, como ocurre con una alta ingesta de Na+ o en el tratamiento con diuréticos del asa, están asociados con una elevada excreción de K.
Determinación de laboratorio
La determinación de laboratorio de la concentración plasmática de K es generalmente exacta. Los métodos más antiguos que utilizaban la fotometría de llama han sido sustituidos en gran parte por la determinación con electrodos específicos de los iones. Se dispone actualmente de técnicas colorimétricas más modernas para la determinación rápida del K plasmático a la cabecera del Paciente. Estas últimas son razonablemente exactas y, aunque no sustituyen a las determinaciones del laboratorio clínico, son de utilidad, especialmente en una UCI, por la rápida disponibilidad de los resultados.
Varios estados patológicos conducen a valores erróneos de la concentración de K. Concentraciones falsamente bajas del K sérico (seudohipopotasemia) se producen a veces en Pacientes de leucemia mieloide con un recuento leucocitario extremadamente elevado (>105/m l) si la muestra de sangre se deja sedimentar a temperatura ambiente antes de ser procesada, debido a la captación de K plasmático por los leucocitos anormales de la muestra. La seudohipopotasemia puede evitarse con una separación inmediata del plasma o el suero en las muestras de sangre obtenidas para la determinación de los electrólitos. También puede producirse una concentración sérica de K falsamente elevada (seudohiperpotasemia), muy frecuentemente causada por hemólisis y salida del K intracelular de los eritrocitos de la muestra. Por este motivo, el personal que realiza la punción venosa debe tener cuidado para no aspirar la sangre con rapidez a través de una aguja de estrecho calibre o de no agitar excesivamente las muestras de sangre. La seudohiperpotasemia puede producirse por una trombocitosis (recuento de plaquetas >106/m l) debida a la liberación de K de las plaquetas durante la coagulación. En los casos de seudohiperpotasemia, el K plasmático (sangre no coagulada), al contrario que el K sérico, será normal.
TRASTORNOS DEL METABOLISMO DEL POTASIO
Hipopotasemia
Disminución de la concentración de potasio sérico por debajo de 3,5 mEq/l, causada por un déficit en los depósitos de potasio corporales totales o por un desplazamiento anormal del potasio al interior de las células.
Etiología y patogenia
La hipopotasemia puede estar causada por una disminución de la ingesta de K, pero habitualmente se debe a pérdidas excesivas de K en la orina o el tracto GI. Las pérdidas gastrointestinales anormales de K se producen en la diarrea crónica e incluyen las debidas al abuso crónico de laxantes o a una derivación intestinal. Otras causas de pérdidas gastrointestinales de K son la pica de yeso, los vómitos y la aspiración gástrica. En raras ocasiones, el adenoma velloso del colon puede causar una pérdida masiva de K por el tracto GI. Las pérdidas gastrointestinales de K pueden complicarse con pérdidas renales de K producidas por alcalosis metabólica y estimulación de la aldosterona debida a la depleción de volumen.
También puede causar hipopotasemia el desplazamiento transcelular de K al interior de las células. Esto puede producirse en la glucogénesis durante la NPT o la hiperalimentación enteral o tras la administración de insulina. La estimulación del sistema nervioso simpático, en especial la de los agonistas b2, como el albuterol o la terbutalina, puede producir hipopotasemia debida a captación celular de K. Análogamente, en Pacientes con tirotoxicosis se presenta a veces hipopotasemia intensa por un exceso de estimulación simpática b-adrenérgica (parálisis periódica tirotóxica hipopotasémica). La parálisis periódica familiar es una rara enfermedad autosómica dominante caracterizada por episodios transitorios de hipopotasemia intensa que se creen debidos a bruscos desplazamientos anormales de K hacia dentro de las células (v. Hiperpotasemia, más adelante). Los episodios se asocian frecuentemente con grados variables de parálisis. Se desencadenan característicamente por una comida abundante en hidratos de carbono o por el ejercicio extenuante, pero se han descrito variantes sin esos rasgos.
Diversos trastornos pueden causar pérdidas excesivas renales de K. Puede producirse caliuresis en el exceso de esteroides suprarrenales debido a los efectos directos de los mineralocorticoides sobre la secreción de K en la nefrona distal. El síndrome de Cushing, el hiperaldosteronismo primario, los raros tumores secretores de renina, el aldosteronismo sensible al tratamiento con glucocorticoides (un raro trastorno heredado) y la hiperplasia suprarrenal congénita pueden causar hipopotasemia por el exceso de formación de mineralocorticoides. La inhibición de la enzima 11-b-hidroxiesteroide deshidrogenasa (11-b-HSDH) evita la conversión del cortisol, que tiene alguna actividad mineralocorticoide, en cortisona, que carece de ella. Sustancias como el ácido glicirretínico (que se encuentra en el regaliz y el tabaco de mascar) inhibe la 11-b -HSDH, produciéndose altos niveles circulantes de cortisol y pérdida excesiva renal de K.
El síndrome de Liddle (v. también cap. 229) es un raro trastorno autosómico dominante caracterizado por hipertensión grave e hipopotasemia. El síndrome de Liddle es causado por una reabsorción de Na+ desenfrenada en la nefrona distal debida a una de las diversas mutaciones detectadas en los genes que codifican las subunidades de canales de Na+ en los epitelios. La reabsorción inadecuadamente alta de Na+ causa tanto hipertensión como derroche renal de K.
El síndrome de Bartter (v. también cap. 229) es un trastorno infrecuente de causa dudosa caracterizado por una pérdida excesiva de K y Na+, producción excesiva de renina y aldosterona y tensión normal.
Finalmente, la pérdida excesiva renal de K puede ser causada por muchas enfermedades congénitas y adquiridas del túbulo renal, como, por ejemplo, la acidosis tubular renal y el síndrome de Fanconi, síndrome infrecuente que causa pérdida renal de K, glucosa, fosfato, ácido úrico y aminoácidos.
Los diuréticos son, con mucho, los fármacos más comúnmente utilizados que producen hipopotasemia. Los diuréticos que pierden K bloquean la reabsorción de Na+ proximal a la nefrona distal; incluyen las tiazidas, los diuréticos del asa y los diuréticos osmóticos. La espironolactona, la amilorida y el triamtereno bloquean la reabsorción de Na+ en el túbulo distal y el conducto colector y no se asocian por ello con pérdida excesiva de K. Al inducir diarrea, los laxantes, especialmente si se abusa de ellos, pueden causar hipopotasemia. El abuso subrepticio de diuréticos y/o laxantes es una causa frecuente de hipopotasemia persistente, particularmente entre los Pacientes obsesionados con la pérdida de peso y entre los profesionales sanitarios que tienen acceso a las medicaciones prescritas.
Otros fármacos que pueden causar hipopotasemia son la anfotericina B, las penicilinas antiseudomónicas (como la carbenicilina) y la penicilina a dosis altas. Por último, la hipopotasemia se ha observado en la intoxicación por teofilina, tanto aguda como crónica.
Síntomas, signos y diagnóstico
La hipopotasemia grave (con un K plasmático <3 mEq/l) puede producir debilidad muscular y conducir a parálisis e insuficiencia respiratoria. Otras disfunciones musculares consisten en calambres, fasciculaciones, íleo paralítico, hipoventilación, hipotensión, tetania y rabdomiólisis. La hipopotasemia persistente puede dificultar la capacidad concentradora del riñón, produciendo poliuria con polidipsia secundaria. Suele existir alcalosis metabólica, aunque la hipopotasemia también se produce en la acidosis metabólica, como en la diarrea o la acidosis tubular renal. Generalmente la hipopotasemia no afecta a la TFG, ni al equilibrio de agua, ni al Na+. Sin embargo, puede producirse un estado parecido a la diabetes insípida nefrógena con grave depleción de K.
Los efectos cardíacos de la hipopotasemia suelen ser mínimos en tanto los niveles plasmáticos de K no sean <3 mEq/l. La hipopotasemia puede producir contracciones ventriculares y auriculares prematuras, taquiarritmias ventriculares y auriculares y bloqueo auriculoventricular de segundo o tercer grado. Los Pacientes con una cardiopatía preexistente importante, y/o los que reciben digital, corren el riesgo de presentar anomalías de la conducción cardíaca incluso por una hipopotasemia muy leve. En la figura 12-1 se muestran los cambios característicos de depresión del segmento ST, aumento de amplitud de la onda U y una onda T de menos amplitud que la onda U (en la misma derivación).
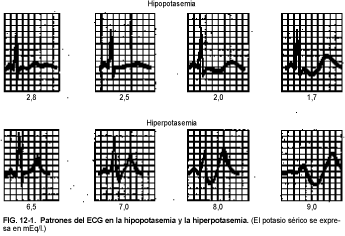
El diagnóstico de hipopotasemia se hace fundándose en un nivel plasmático o sérico de K <3,5 mEq/l (v. Metabolismo del potasio, más atrás).
Profilaxis y tratamiento
En la mayoría de los Pacientes que reciben diuréticos no es imprescindible la reposición rutinaria del K. Sin embargo, evitar la hipopotasemia es particularmente importante en los Pacientes que reciben digital, en los Pacientes asmáticos que reciben agonistas ß2 y en los diabéticos no dependientes de insulina. Estos Pacientes deben recibir la mínima dosis efectiva de un diurético con una duración de acción moderada; se debe restringir la ingesta dietética de Na+ (<2 g/d) y monitorizar estrictamente el K plasmático tras la iniciación del tratamiento. Una vez comprobada una concentración de K estable, se necesita una monitorización menos frecuente, a no ser que se aumente la dosis o aparezcan síntomas de hipopotasemia u otros problemas. Si se presenta hipopotasemia está indicada la suplementación con K y el diurético debe interrumpirse si es posible. La adición de 100 m g/d de triamtereno o de 25 mg de espironolactona 4/d puede ser útil en algunos Pacientes que se hacen hipopotasémicos con el tratamiento diurético, pero debe evitarse en Pacientes con insuficiencia renal, diabetes u otra enfermedad renal intersticial asociada con hipopotasemia debida a hipoaldosteronismo hiporreninémico (acidosis tubular renal tipo 4). La deficiencia de K debe corregirse con gran cuidado en los Pacientes con insuficiencia renal.
La corrección de la causa subyacente puede bastar cuando la hipopotasemia es leve. Cuando el déficit y la hipopotasemia son más intensos (K plasmático <3 mEq/l), o cuando es imprescindible un tratamiento continuado con agentes que producen depleción de K, se puede administrar KCl v.o. (cloruro potásico al 10%). Generalmente, 20 a 80 mEq/d, además de las pérdidas actuales de K, administrados en dosis fraccionadas a lo largo de varios días, corrigen el déficit de K. Por otra parte, la necesidad de los suplementos de K puede continuar durante varias semanas en el caso de la realimentación tras una inanición prolongada.
Se dispone de diversos suplementos orales de K. El cloruro potásico en solución administrado por v.o. se tolera mal en dosis superiores a 25 a 50 mEq a causa de su sabor amargo. Se ha descrito que los preparados de K con cubierta entérica producen ulceración del intestino delgado. Los preparados de cloruro potásico impregnados con cera parecen ser inocuos y se toleran bien. El sangrado GI puede ser aún menos frecuente con los preparados de cloruro potásico microencapsulado. Se dispone de varios preparados que contienen 8 o 10 mEq/cápsula.
Cuando la hipopotasemia es intensa y produce síntomas, o no responde al tratamiento oral, es preciso reponer el K por vía parenteral. El ritmo de corrección de la hipopotasemia está limitado por la demora en la disponibilidad del potasio en su transferencia hacia las células. En los casos de déficit de K con una concentración de K plasmático alta, como en la cetoacidosis diabética, es preciso esperar hasta que el K plasmático empiece a disminuir antes de administrar K por vía i.v. Aun cuando el déficit de K sea intenso, rara vez es necesario administrar >80 a 100 mEq de potasio sobre las pérdidas continuadas en un período de 24 h. Las modernas y precisas bombas de infusión i.v. han reducido el riesgo de administrar soluciones de KCl sumamente concentradas. Por lo demás, en la mayoría de las situaciones no es preciso que las concentraciones de potasio de las soluciones i.v. sean mayores de 60 mEq/l, y las tasas de infusión no deben exceder los 10 mEq/h. A veces puede ser necesario administrar soluciones de KCl i.v. con más rapidez para evitar una hipopotasemia grave y progresiva. La infusión de >40 mEq/h de cloruro potásico sólo debe llevarse a cabo con monitorización cardíaca continua y determinaciones horarias del K plasmático para evitar una hiperpotasemia grave y/o una parada cardíaca. Las soluciones de glucosa no son una opción ideal para administrar KCl, porque la elevación subsiguiente del nivel de insulina plasmática del Paciente podría conducir a un empeoramiento transitorio de la hipopotasemia con agravamiento de los síntomas, especialmente en los Pacientes digitalizados. Finalmente, cuando la hipopotasemia se asocia a hipomagnesemia, generalmente es necesario corregir la deficiencia de magnesio, detener la pérdida renal de K y facilitar su reposición (v. Hipomagnesemia, más adelante).
Hiperpotasemia
Aumento de la concentración de potasio sérico por encima de 5,5 mEq/l (potasio plasmático superior a 5,0) causada por un exceso de los depósitos de potasio corporal total o un movimiento anormal de salida de potasio de las células.
Etiología y patogenia
Dado que los riñones excretan normalmente las cargas de K en función del tiempo, una hiperpotasemia sostenida suele implicar una disminución de la excreción renal de K. La hiperpotasemia también puede ser causada por el movimiento transcelular del K hacia fuera de las células en estados de acidosis metabólica, hiperglucemia en presencia de una deficiencia de insulina, ejercicio moderadamente intenso, especialmente en presencia de un bloqueo ß, intoxicación digitálica, lisis tumoral aguda, hemólisis intravascular aguda o rabdomiólisis. La parálisis periódica familiar hiperpotasémica es un raro trastorno heredado debido a una salida brusca del K de las células desencadenado generalmente por el ejercicio.
La hiperpotasemia por un exceso del K corporal total es particularmente frecuente en los estados oligúricos (en especial en la insuficiencia renal aguda) y se asocia con la rabdomiólisis, las quemaduras, las hemorragias en los tejidos blandos o el tracto GI y con la insuficiencia suprarrenal, la cual se identifica cada vez más en los Pacientes con SIDA (v. más atrás, Hiponatremia). En la insuficiencia renal crónica, la hiperpotasemia es infrecuente hasta que la TFG desciende por debajo de 10 a 15 ml/min, salvo si la ingesta dietética de K es excesiva o existe otra fuente de exceso de carga de K, como un tratamiento con K oral o parenteral, hemorragia GI, lesión de los tejidos o hemólisis. Otras causas posibles de hiperpotasemia en la insuficiencia renal crónica son el hipoaldosteronismo hiporreninémico (acidosis tubular renal tipo 4), los inhibidores de la ECA, los diuréticos ahorradores de K, el ayuno (supresión de la secreción de insulina), los fármacos bloqueantes ß y los AINE. Si se ingiere suficiente KCl por vía oral o se administra por vía parenteral, puede producirse una hiperpotasemia grave incluso con una función renal normal. No obstante, la hiperpotasemia yatrogénica se observa con más frecuencia en Pacientes con algún grado de deterioro renal. Otros fármacos que pueden limitar la eliminación renal de K, y producir con ello una hiperpotasemia, son la ciclosporina, el litio, la heparina y la trimetoprima.
Síntomas, signos y diagnóstico
Aunque a veces se produce parálisis fláccida, la hiperpotasemia suele ser asintomática hasta que sobreviene la toxicidad cardíaca (v. fig. 12-1). Los primeros cambios observados en el ECG con una hiperpotasemia progresiva (K plasmática >5,5 mEq/l) son un acortamiento del intervalo QT y ondas T altas, simétricas y afiladas. La hiperpotasemia progresiva (K plasmático >6,5 mEq/l) produce arritmias nodales y ventriculares, ensanchamiento del complejo QRS, alargamiento del intervalo PR y desaparición de la onda P. Finalmente, el complejo QRS degenera a un patrón de onda sinusal y se inicia la asistolia o la fibrilación.
En la parálisis periódica familiar hiperpotasémica aparece frecuentemente debilidad durante los ataques y puede evolucionar a una parálisis manifiesta.
El diagnóstico de hiperpotasemia se hace con un nivel plasmático o sérico de K >5,5 mEq/l (en plasma >5,0 mEq/l) (v. más atrás, Metabolismo del potasio).
Tratamiento
La hiperpotasemia leve (con un K plasmático <6 mEq/l) puede responder a una ingesta de K reducida o a la interrupción de fármacos como los diuréticos ahorradores de K, los bloqueantes b, los AINE o los inhibidores de la ECA. La adición de un diurético del asa puede potenciar también la excreción renal de K. Un K plasmático >6 mEq/l exige un tratamiento más intervencionista. Sin embargo, en la insuficiencia renal aguda o crónica, especialmente en presencia de hipercatabolismo o de lesión tisular, el tratamiento se debe iniciar cuando el nivel de K plasmático supera los 5 mEq/l.
Si no existen anomalías en el ECG y el K plasmático no está muy aumentado (<6 mEq/l), se puede administrar sulfonato sódico de poliestireno en sorbitol (15 a 30 g en 30 a 70 ml de sorbitol al 70% v.o. cada 4 a 6 h). El sulfonato Na+ de poliestireno actúa como una resina de intercambio catiónico y elimina el K a través de la mucosa GI. El sorbitol se administra con la resina para asegurar el tránsito a través del tracto GI. Los Pacientes que no pueden tomar medicamentos orales debido a íleo u otros motivos pueden recibir dosis similares mediante un enema de retención. Se elimina alrededor de 1 mEq de K por gramo de resina administrado. El tratamiento con resina actúa con lentitud y a menudo no logra reducir el K plasmático de manera importante en los estados hipercatabólicos. Dado que el K se intercambia por Na+ cuando se emplea el sulfonato Na+ de poliestireno, puede presentarse una sobrecarga de Na+, sobre todo en Pacientes oligúricos con sobrecarga preexistente de la volemia.
En urgencias como la toxicidad cardíaca o si el nivel plasmático de K es >6 mEq/l, deben llevarse a cabo inmediatamente las tres medidas que siguen en sucesión rápida sin esperar a repetir las cifras plasmáticas de K después de cada una de ellas:
1. Administración i.v. de 10 a 20 ml de gluconato de calcio al 10% (o de 5 a 10 ml de gluceptato cálcico al 22%) a lo largo de 5 a 10 minutos. Se debe tener cuidado al administrar calcio a Pacientes que toman digital, dado el riesgo de desencadenar arritmias relacionadas con la hipopotasemia. Si el ECG ha empeorado hasta la aparición de ondas sinusales o de asistolia, el gluconato cálcico puede administrarse en infusión i.v. rápida (5 a 10 ml en 2 min).
2. Administración i.v. de 5 a 10 U de insulina regular en forma de pulso i.v. seguido inmediatamente por la infusión rápida de 50 ml de glucosa al 50%. Esto debe ir seguido de solución de dextrosa al 10% a 50 ml/h para evitar la hipoglucemia. El efecto sobre el K plasmático aparece en 15 min.
3. Inhalación de una dosis alta de un agonista ß, como el albuterol (10 a 20 mg) a lo largo de 10 min (concentración 5 mg/ml). Esto ha demostrado ser eficaz y seguro para tratar la hiperpotasemia. El comienzo de la acción tiene lugar antes de unos 30 min. La duración del efecto es de 2 a 4 h.
Nota: En este algoritmo se ha omitido a propósito el Na+HCO3. Recientemente se ha cuestionado la eficacia de la administración empírica de Na+HCO3 para el tratamiento de la hiperpotasemia aguda que pone en peligro la vida del Paciente.
Además de las estrategias ya mencionadas para reducir el K mediante su desplazamiento hacia las células, las medidas para eliminar K del organismo deben llevarse a cabo al comenzar el tratamiento de la hiperpotasemia grave o con síntomas. La eliminación de K puede realizarse a través del tracto GI mediante la administración del sulfonato sódico de poliestireno o mediante hemodiálisis. Esta última debe instaurarse con prontitud, tras las medidas de urgencia, en los Pacientes con insuficiencia renal o si el tratamiento de urgencia es ineficaz. La diálisis peritoneal es relativamente ineficiente para la eliminación del K, pero puede ser beneficiosa para el Paciente con acidosis, en especial si es probable que se presente una sobrecarga de volumen con el Na+HCO3.
METABOLISMO DEL CALCIO
El calcio (Ca) es necesario para el funcionamiento correcto de numerosos procesos intracelulares y extracelulares, como la contracción muscular, la conducción nerviosa, la liberación de hormonas y la coagulación sanguínea. Además, el ion Ca representa un papel singular en la señalización intracelular y está implicado en la regulación de muchas enzimas. Por consiguiente, es esencial mantener la homeostasis del Ca.
Las concentraciones de Ca, tanto extracelulares como intracelulares, están estrechamente reguladas por el transporte bidireccional de Ca a través de la membrana plasmática de las células y por las membranas de orgánulos intracelulares como las del retículo endoplasmático, las del retículo sarcoplásmico de las células musculares y las de las mitocondrias. El transporte de Ca fuera del citoplasma de las células hacia esos diversos compartimientos y el alto grado de unión del Ca a las proteínas mantiene la concentración de Ca ionizado en el citoplasma en el intervalo micromolar (es decir, menor que 1/1.000 de la concentración de Ca plasmático). Dado que el Ca existe en el citosol en concentraciones tan bajas, es singularmente apropiado para actuar como un segundo mensajero intracelular. En el músculo esquelético, los aumentos transitorios en la concentración citosólica de Ca originan la interacción entre el Ca y las proteínas fijadoras de Ca, troponina C y calmodulina, e inician la contracción muscular. El acoplamiento excitación-contracción en el músculo cardíaco y en el músculo liso también es Ca-dependiente. La concentración intracelular de Ca regula otros procesos celulares diferentes mediante la acción de proteincinasas y la fosforilación de enzimas. El Ca está también involucrado en la acción de otros mensajeros intracelulares, como el adenosina monofosfato cíclico (AMPc) y el inositol 1,4,5-trifosfato, y de esta forma es mediador de la respuesta celular a numerosas hormonas, como adrenalina, glucagón, vasopresina, secretina y colecistocinina.
A pesar de sus importantes funciones intracelulares, en torno al 99% del Ca corporal está en el hueso, donde se encuentra sobre todo constituyendo complejos con otros iones en forma de cristales de hidroxiapatita. Aproximadamente un 1% del Ca óseo es libremente intercambiable con el LEC y, por consiguiente, está disponible para amortiguar los cambios en el equilibrio del Ca. Los niveles normales de Ca plasmático total oscilan desde 8,8 a 10,4 m g/dl (2,20 a 2,60 m mol/l). Alrededor del 40% del Ca total en la sangre está unido a las proteínas plasmáticas, principalmente a la albúmina. El 60% restante comprende el Ca ionizado y el Ca en forma de complejo con fosfato y citrato.
El Ca total (es decir, el Ca unido a proteínas, el Ca en forma de complejo y el Ca ionizado) es el que suele determinarse en el laboratorio clínico como Ca plasmático. En un caso ideal debería determinarse el Ca ionizado o el Ca libre, puesto que ésta es la forma fisiológicamente activa del Ca en el plasma. Se sabe que el Ca ionizado ha sido siempre difícil de medir y todavía no se determina de manera rutinaria. Sin embargo, el Ca ionizado puede medirse actualmente con electrodos ion-específicos, lo que es útil a veces en Pacientes en los que se sospechan alteraciones importantes de la unión del Ca plasmático a las proteínas.
La fracción de Ca ionizado se valora por lo general a partir del Ca plasmático total. El Ca ionizado es aproximadamente un 50% del Ca plasmático total y varía de 4,7 a 5,2 m g/dl (1,17 a 1,3 m mol/l). En la unión del Ca a las proteínas influyen los cambios del pH. La acidosis se asocia con un aumento del Ca ionizado debido a la disminución de la unión a proteínas, mientras que la alcalosis se asocia con disminución de Ca ionizado por un aumento de unión a las proteínas. Las alteraciones de la concentración de proteínas plasmáticas influyen también sobre la fracción de Ca ionizado. En la hipoalbuminemia, por ejemplo, la determinación del Ca plasmático suele estar baja, pero dado que ésta refleja sobre todo los niveles bajos de la fracción de Ca unido a proteínas, el Ca ionizado puede estar normal. La concentración de Ca plasmático total puede corregirse en función del nivel de albúmina utilizando la aproximación de que el Ca plasmático total medido disminuirá o aumentará 0,8 m g/dl (0,20 m mol/l) por cada g/dl de disminución o aumento de la albúmina. Así, un Paciente con una albúmina de 2,0 g/dl (normal, 4,0) tendría una reducción en el Ca plasmático medido de 1,6 m g/dl (2 x 0,8 m g/dl) sólo por la hipoalbuminemia. En este caso, los límites del intervalo de los valores de Ca plasmático aceptables se reducirían en 1,6 m g/dl (0,40 m mol/l); alternativamente, el Ca plasmático del Paciente puede aumentarse con el mismo factor y compararse con los intervalos normales para el Ca plasmático citados antes. Por desgracia, este y otros algoritmos para predecir el Ca ionizado suelen ser inexactos. En consecuencia, el Ca plasmático ionizado debe determinarse directamente siempre que se sospeche una anomalía del Ca ionizado, aunque exista un Ca plasmático total normal. Los aumentos de las proteínas plasmáticas, como ocurre en el mieloma múltiple, elevan el Ca plasmático total a expensas de la fracción unida a las proteínas. Dado que las paraproteínas fijan el Ca plasmático de forma imprevisible, para orientar las decisiones clínicas se debe medir directamente el Ca plasmático ionizado.
El mantenimiento de los depósitos de Ca del organismo y de la concentración plasmática de Ca dependen en último término de la ingesta dietética de Ca, de la absorción de Ca en el tracto GI y de la excreción renal de Ca. En una dieta equilibrada se ingieren unos 1.000 mg diarios de Ca. Alrededor de 200 m g/d se pierden en la luz del tracto GI con la bilis y otras secreciones. En función de la concentración de 1,25-dihidroxivitamina D, de 200 a 400 mg de Ca se absorben diariamente a partir del intestino. Los 800 a 1.000 mg restantes (20 a 25 mmol) aparecen en las heces. El equilibrio neto del Ca se mantiene mediante la excreción renal del Ca, que es en promedio de 200 m g/d (5 mmol/d).
Regulación del metabolismo del calcio
El metabolismo del Ca y el fosfato (PO4-, v. más adelante) están íntimamente relacionados. La regulación del Ca y el fosfato está influida considerablemente por los niveles circulantes de hormona paratiroidea (PTH), vitamina D y, en menor medida, por la hormona calcitonina. Las concentraciones de Ca y fosfato inorgánico también se relacionan por la capacidad de ambos para reaccionar químicamente entre sí para formar fosfato cálcico. Se calcula que el producto de las concentraciones de Ca y PO4 (en mEq/l) in vivo es normalmente 60. Cuando el producto de solubilidad de Ca y PO4 es mayor de 70, el riesgo de precipitación de cristales de fosfato cálcico en los tejidos blandos está muy aumentado. La precipitación en el tejido vascular es especialmente preocupante, porque puede conducir a una enfermedad vascular arteriosclerótica precipitada.
La PTH es un polipéptido de 84 aminoácidos secretado por las glándulas paratiroides. Tiene varias acciones, pero quizá la más importante es la defensa contra la hipocalcemia. Las células paratiroideas detectan los descensos del Ca plasmático, presumiblemente por medio de un receptor de Ca, e incrementan la expresión del gen de la PTH y la liberación de PTH preformada hacia la circulación. El Ca plasmático aumenta al cabo de minutos debido a varias acciones de la PTH, como aumentar la absorción renal e intestinal de Ca y movilizar rápidamente Ca y PO4 a partir del hueso (reabsorción ósea). La excreción renal de Ca es generalmente paralela a la excreción de Na+ y está influida por los mismos factores que regulan el transporte de Na+ en el túbulo proximal. Sin embargo, la PTH potencia la reabsorción de Ca en el túbulo distal independientemente del Na+. La PTH reduce también la reabsorción renal de PO4 y aumenta así las pérdidas renales de PO4. Estas pérdidas evitan que se sobrepase la solubilidad del fosfato cálcico en el plasma cuando los niveles de Ca aumentan en respuesta a la PTH.
La PTH influye también sobre el metabolismo de la vitamina D. La vitamina D incrementa el porcentaje de Ca dietético absorbido en el intestino. La enzima 1-a-hidroxilasa convierte a la vitamina D en su forma más activa, 1,25-dihidroxicolecalciferol [1,25(OH)2D3] en el riñón. La 1,25(OH)2D3 facilita el transporte de Ca en el intestino, en parte porque media en la síntesis de proteínas fijadoras de Ca de la mucosa. La PTH potencia la absorción de Ca intestinal por medio de un aumento en la formación de 1,25(OH)2D3. A pesar de este efecto sobre la absorción de Ca a partir del tracto GI, los aumentos prolongados de la secreción de PTH conducen generalmente a una nueva reabsorción ósea al inhibir la función de los osteoblastos y estimular la actividad de los osteoclastos. Ambas hormonas funcionan in vivo como importantes reguladores del crecimiento óseo y de la remodelación ósea (v. Deficiencia y dependencia de vitamina D, cap. 3).
La exploración de la función paratiroidea incluye la determinación de los niveles circulantes de PTH mediante radioinmunoensayo y la determinación de la excreción de AMPc total y nefrógeno en la orina. En la actualidad ya apenas se mide el AMPc urinario, ya que se dispone de forma generalizada de ensayos exactos de la PTH. Deben elegirse siempre los ensayos de la molécula de PTH intacta con preferencia sobre los demás. Los antiguos ensayos del extremo carboxiterminal eran difíciles de interpretar en Pacientes con insuficiencia renal.
La calcitonina es una hormona polipeptídica de 32 aminoácidos secretada por las células C tiroideas. La calcitonina tiende a disminuir la concentración plasmática de Ca facilitando la captación celular, la excreción renal y la formación de hueso. Los efectos de la calcitonina sobre el metabolismo óseo son mucho más débiles que los de la PTH o la vitamina D.
TRASTORNOS DEL METABOLISMO DEL CALCIO
Hipocalcemia
Disminución de la concentración de calcio plasmático total por debajo de 8,8 m g/dl (2,20 m mol/l) en presencia de una concentración normal de proteínas plasmáticas.
La hipocalcemia en los recién nacidos se expone en Problemas metabólicos en el recién nacido, capítulo 260.
Etiología y patogenia
La hipocalcemia tiene una serie de causas. Algunas se enumeran a continuación:
1. El hipoparatiroidismo es el resultado de una deficiencia o ausencia de PTH. Se caracteriza por hipocalcemia e hiperfosfatemia y suele asociarse con tetania crónica. El hipoparatiroidismo suele ser consecuencia de la extirPación accidental o de la lesión de varias glándulas paratiroides durante la tiroidectomía. El hipoparatiroidismo transitorio es frecuente después de una tiroidectomía subtotal. El hipoparatiroidismo permanente se produce en menos del 3% de las tiroidectomías realizadas por manos expertas. Las manifestaciones de la hipocalcemia suelen iniciarse unas 24 a 48 h después de la operación, pero pueden presentarse por primera vez tras varios meses o años. La deficiencia de PTH es más frecuente tras una tiroidectomía radical por cáncer o como consecuencia de una operación sobre las propias paratiroides (paratiroidectomía subtotal o total). Los factores de riesgo de una hipocalcemia grave tras una paratiroidectomía subtotal son la hipercalcemia preoperatoria, la extirPación quirúrgica de un adenoma voluminoso y una fosfatasa alcalina elevada.
El hipoparatiroidismo idiopático es una enfermedad poco frecuente en la cual las glándulas paratiroides están ausentes o atrofiadas. Puede aparecer esporádicamente, o bien como un trastorno hereditario. Las glándulas paratiroides faltan a veces, en asociación con aplasia del timo y anomalías de las arterias que se originan en los arcos branquiales (síndrome de DiGeorge). Otras formas hereditarias son el síndrome de hipoparatiroidismo genético ligado al cromosoma X, la enfermedad de Addison y la candidiasis mucocutánea (v. cap. 11).
El seudohipoparatiroidismo constituye un grupo de trastornos caracterizados, no por deficiencia de PTH, sino por resistencia del órgano diana a su acción. Se han descrito formas autosómicas dominantes así como recesivas. Los Pacientes con seudohipoparatiroidismo tipo I (osteodistrofia hereditaria de Albright) suelen estar hipocalcémicos, pero pueden tener concentraciones plasmáticas de Ca normales.
El sello característico de este síndrome es una insuficiencia del riñón para desarrollar la respuesta fosfatúrica normal o el aumento del AMPc urinario tras la administración de PTH. La orina contiene AMPc filtrado y también AMPc generado en el riñón. El ajuste del AMPc urinario para el componente no nefrogénico conduce a una medida más específica del efecto de la PTH sobre el riñón. Por ello, la producción nefrogénica de AMPc permite determinar mejor si existe una respuesta correcta a la PTH. Muchos de estos Pacientes presentan una deficiencia de la proteína reguladora GSa, estimuladora de la unión al nucleótido de guanina. En familias con seudohipoparatiroidismo tipo I se han encontrado mutaciones del gen que codifica esta importante proteína mensajera intracelular. Las anomalías asociadas son: baja estatura, facies redonda, retraso mental con calcificación de los ganglios basales, huesos metacarpianos y metatarsianos acortados, así como un hipotiroidismo leve y otras anomalías endocrinas poco perceptibles. Algunos miembros de las familias afectadas con mutaciones en la proteína GSa tienen muchos de los rasgos de la enfermedad, pero no tienen hipoparatiroidismo, un trastorno que se describe a veces como seudo-seudohipoparatiroidismo. El seudohipoparatiroidismo tipo II es aún menos frecuente. En estos Pacientes, la PTH no eleva el Ca plasmático o el PO4 urinario, pero la respuesta del AMPc nefrogénico a la PTH exógena es normal.
2. La deficiencia de vitamina D es una causa importante de hipocalcemia. La deficiencia de vitamina D puede ser consecuencia de una ingesta dietética insuficiente o de una disminución de la absorción debida a enfermedad hepatobiliar o a malabsorción intestinal. También puede producirse por alteraciones en el metabolismo de la vitamina D, como ocurre con ciertos fármacos (difenilhidantoína, fenobarbital y rifampicina) o por falta de exposición de la piel a la luz solar. Esta última es una causa importante de deficiencia adquirida de vitamina D en climas nórdicos, entre los individuos que llevan vestidos que los cubren completamente (p. ej., mujeres musulmanas en Inglaterra). El raquitismo de tipo I dependiente de vitamina D es un trastorno autosómico recesivo en el cual existe una deficiencia de la enzima 1-a-hidroxilasa necesaria para convertir 25(OH)D3 en 1,25(OH)2D3. En el raquitismo de tipo II dependiente de vitamina D, los órganos diana no pueden responder a la 1,25(OH)2D3. La deficiencia de vitamina D está asociada con hipocalcemia e hipofosfatemia intensa. Puede producirse debilidad y dolor muscular y deformidades óseas típicas (v. Deficiencia y dependencia de la vitamina D, cap. 3).
3. La enfermedad tubular renal -incluido el síndrome de Fanconi debido a nefrotoxinas como los metales pesados y la acidosis tubular renal- puede causar una grave hipocalcemia debida a la pérdida anormal renal de Ca y a una disminución de la conversión renal a vitamina D. El cadmio, en particular, causa hipocalcemia por lesión de las células del túbulo proximal y por interferencia en la conversión a vitamina D. También puede producirse osteomalacia debida a la enfermedad tubular renal, pero es posible que se deba principalmente a la acidosis crónica asociada.
4. La insuficiencia renal puede conducir a hipocalcemia por disminución de formación de 1,25(OH)2D3 a causa de daño celular renal directo, así como por la hiperfosfatemia debida a la reducción de la secreción renal de fosfato.
5. La depleción de magnesio presente en la malabsorción intestinal o en la deficiencia dietética puede causar hipocalcemia. La deficiencia relativa de PTH y la resistencia del órgano terminal a su acción que aparece en la depleción de magnesio conduce a concentraciones plasmáticas <1,0 mEq/l (<0,5 m mol/l); la repleción del magnesio aumenta los niveles de PTH y la conservación renal del Ca.
6. La pancreatitis aguda causa hipocalcemia cuando el Ca resulta quelado por los productos lipolíticos liberados desde el páncreas inflamado.
7. La hipoproteinemia de cualquier causa puede reducir la fracción plasmática de Ca unido a las proteínas. La hipocalcemia debida a una disminución de la unión a las proteínas es asintomática. Dado que la fracción de Ca ionizado no se altera, esta entidad se ha denominado hipocalcemia facticia.
8. La formación aumentada de hueso con una ingesta de Ca insuficiente puede causar hipocalcemia. Esta situación existe particularmente tras la corrección quirúrgica del hiperparatiroidismo en los Pacientes con osteítis fibrosa quística grave y se ha denominado síndrome del hueso hambriento.
9. El shock séptico puede asociarse con una hipocalcemia debida a la supresión de la liberación de PTH y de la conversión de 25(OH)D3 a 1,25(OH)2D3.
10. La hiperfosfatemia causa también hipocalcemia por uno o varios mecanismos insuficientemente conocidos. Pacientes con insuficiencia renal y retención subsiguiente de fosfato son particularmente propensos a esta forma de hipocalcemia.
11. Los fármacos asociados con hipocalcemia son los utilizados generalmente para tratar la hipercalcemia (v. Hipercalcemia, más adelante); los anticonvulsivos (difenilhidantoína, fenobarbital) y la rifampicina, que alteran el metabolismo de la vitamina D; la transfusión de productos sanguíneos tratados con citrato y también los agentes de contraste radiográfico que contienen el agente quelante iónico divalente etilendiaminotetraacetato.
12. Aunque sería de esperar que una secreción excesiva de calcitonina pudiera causar hipocalcemia, se producen rara vez niveles bajos de Ca plasmático en Pacientes con grandes cantidades circulantes de calcitonina procedentes de un carcinoma medular del tiroides.
Síntomas y signos
La hipocalcemia es frecuentemente asintomática. La presencia de hipoparatiroidismo la sugieren a menudo las manifestaciones clínicas de una enfermedad subyacente (p. ej., cataratas, calcificación de los ganglios basales y candidiasis crónica en el hipoparatiroidismo idiopático).
Las manifestaciones clínicas de hipocalcemia se deben a una alteración del potencial de la membrana celular. Los síntomas se producen principalmente por la irritabilidad neuromuscular. Los calambres musculares que afectan a la espalda y a las piernas son una queja frecuente en Pacientes con hipocalcemia. Una hipocalcemia insidiosa y de desarrollo lento puede producir una leve encefalopatía difusa y debe sospecharse en cualquier Paciente con demencia, depresión o psicosis no explicadas. Tras una hipocalcemia prolongada puede desarrollarse a veces papiledema y cataratas. Una hipocalcemia intensa con un Ca plasmático <7 m g/dl (<1,75 m mol/l) puede causar tetania, laringospasmo o convulsiones generalizadas.
La tetania se produce característicamente por una hipocalcemia grave. También puede producirse por una reducción de la fracción ionizada del Ca plasmático sin una hipocalcemia intensa, como ocurre en la alcalosis grave. La tetania se caracteriza por síntomas sensitivos que consisten en parestesias de los labios, la lengua, las manos y los pies, espasmo carpopedal, que puede ser prolongado y doloroso, molestias musculares generalizadas y espasmo de la musculatura facial. La tetania puede ser manifiesta, con síntomas espontáneos, o latente, que requiere pruebas de provocación para desencadenarse. La tetania latente existe generalmente a concentraciones de Ca plasmático disminuidas menos intensamente: 7 a 8 m g/dl (1,75 a 2,20 m mol/l).
Los signos de Chvostek y Trousseau se exploran con facilidad a la cabecera del Paciente para poner de manifiesto la tetania latente. El signo de Chvostek es una sacudida involuntaria de los músculos faciales provocada por un golpe ligero sobre el nervio facial inmediatamente por delante del conducto auditivo externo. Está presente hasta en un 10% de individuos sanos y suele estar ausente en la hipocalcemia crónica. El signo de Trousseau es el desencadenamiento de un espasmo carpopedal por reducción del aporte sanguíneo a la mano con un torniquete o un manguito de PA inflado 20 mm Hg por encima de la PA sistólica aplicado al antebrazo durante 3 min. El signo de Trousseau aparece también en estados de alcalosis, hipomagnesemia, hipopotasemia e hiperpotasemia, y aproximadamente en un 6% de personas sin alteraciones electrolíticas identificables. La tetania latente puede hacerse manifiesta con una nueva reducción del Ca ionizado por hiperventilación o por la administración de Na+HCO3 o diuréticos que producen depleción de Ca, como la furosemida. Una hipopotasemia concomitante puede enmascarar todas las manifestaciones de la tetania por hipocalcemia.
En Pacientes con hipocalcemia intensa a veces aparecen arritmias o bloqueo cardíaco. El ECG en la hipocalcemia muestra típicamente una prolongación de los intervalos QTC y ST. También pueden observarse cambios en la repolarización, como aguzamiento o inversión de la onda T.
Con la hipocalcemia crónica se asocian otras muchas anomalías, como piel seca y escamosa, uñas quebradizas y pelo áspero. Se presentan a veces infecciones por Candida en la hipocalcemia, pero con mayor frecuencia en Pacientes con hipoparatiroidismo idiopático. Las cataratas oculares se observan a veces en casos de hipocalcemia de larga duración, pero no son reversibles con la corrección del Ca plasmático.
Diagnóstico
La hipocalcemia se diagnostica con un nivel de Ca plasmático total <8,8 m g/dl (<2,20 m mol/l). Cuando existe tetania, el Ca plasmático total suele ser £7 m g/dl (£1,75 m mol/l), a no ser que exista alcalosis. Las anomalías características de la alcalosis metabólica y respiratoria se comentan más adelante en Metabolismo acidobásico. La deficiencia de PTH se caracteriza por Ca plasmático bajo, PO4 plasmático alto y fosfatasa alcalina normal. Aunque en el hipoparatiroidismo el Ca urinario es bajo, resulta relativamente alto si se tiene en cuenta la carga filtrada de Ca en orina.
Dado que la hipocalcemia es el principal estímulo de la secreción de PTH, ésta debería estar elevada en la hipocalcemia. Sin embargo, en el hipoparatiroidismo, la PTH intacta es inadecuadamente baja para el nivel de Ca plasmático. Con menor frecuencia, la PTH es indetectable e indica el diagnóstico de hipoparatiroidismo idiopático, el cual se hace manifiesto en la infancia y puede asociarse con enfermedad de Addison, esteatorrea y candidiasis (v. cap. 11). Existe hiperfosfatemia cuando la hipocalcemia es consecuencia de un hipoparatiroidismo o una insuficiencia renal. Las dos enfermedades se diferencian fácilmente por la presencia de azoemia intensa en la insuficiencia renal.
El seudohipoparatiroidismo tipo I se puede distinguir por la presencia de hipocalcemia a pesar de unos niveles normales a elevados de PTH circulante. El diagnóstico puede sospecharse ante la frecuente existencia de varias anomalías esqueléticas coexistentes, como baja estatura y metacarpianos primero, cuarto y quinto acortados. Los Pacientes con seudohipoparatiroidismo carecen típicamente de las respuestas renales normales a la PTH. A pesar de la presencia de PTH circulante, no existe fosfaturia. Las pruebas de provocación mediante inyección de extracto de paratiroides o de la PTH recombinante humana, pronto disponible, no elevan el AMPc plasmático o nefrogénico. En el seudohiperparatiroidismo tipo II, el extracto de paratiroides eleva el AMPc nefrogénico, pero no induce fosfaturia ni eleva la concentración de Ca plasmático. Antes de hacer el diagnóstico de seudohipoparatiroidismo de tipo II es preciso descartar la deficiencia de vitamina D.
En la osteomalacia o el raquitismo pueden existir las anomalías típicas. El nivel de PO4 plasmático suele estar moderadamente reducido y la fosfatasa alcalina está elevada, reflejando el aumento de movilización de Ca a partir del hueso. La determinación de la 25(OH)D3 y la 1,25(OH)2D3 pueden ayudar a distinguir entre la deficiencia de vitamina D y los estados de dependencia de la vitamina D. El raquitismo hipofosfatémico familiar se identifica por la pérdida renal de PO4 asociada.
Tratamiento
La tetania hipocalcémica grave aguda se trata inicialmente con infusión i.v. de sales de Ca. Puede administrarse gluconato cálcico i.v., 10 ml de solución al 10% a lo largo de 10 min. La respuesta puede ser espectacular, pero en ocasiones sólo dura unas pocas horas. Puede ser necesario repetir las infusiones o añadir una infusión continua con 20 a 30 ml de gluconato cálcico al 10% en 1 litro de solución de dextrosa al 5% a lo largo de las 12 a 24 h siguientes. Las infusiones de Ca son peligrosas en los Pacientes que reciben digital y deben administrarse con lentitud y sólo con monitorización ECG continua. El cloruro cálcico no debe utilizarse si se dispone de alternativas como el gluconato Ca, porque el primero puede causar tromboflebitis grave y necrosis tisulares si se extravasa. Igualmente está contraindicada la inyección i.m. de cualquier preparado de calcio, debido a la necrosis tisular que produce. Cuando la tetania se debe a hipomagnesemia, puede responder pasajeramente a la administración de Ca o K, pero sólo se alivia permanentemente con la repleción de Mg (v. Hipomagnesemia, más adelante).
En el hipoparatiroidismo transitorio tras la tiroidectomía o la paratiroidectomía parcial, el Ca oral suplementario puede ser suficiente para evitar la hipocalcemia. Sin embargo, ésta puede ser particularmente grave y prolongada después de una paratiroidectomía subtotal en los Pacientes con insuficiencia renal crónica o una nefropatía en fase terminal. Puede ser imprescindible una administración parenteral prolongada de Ca para evitar una hipocalcemia grave en el postoperatorio; tal vez haya que suplementar hasta con 1 g de Ca elemento/d durante 5 a 10 d antes de que el Ca y la vitamina D por v. o. sean bastantes para mantener niveles suficientes de Ca en la sangre. En estas situaciones, una fosfatasa alcalina plasmática elevada puede ser un signo de captación rápida de Ca hacia el hueso. Suelen ser precisas grandes cantidades de Ca parenteral hasta que los niveles de fosfatasa alcalina empiecen a disminuir.
En la hipocalcemia crónica generalmente basta con administrar Ca y suplementos ocasionales de vitamina D por v. o. El Ca se puede dar en forma de gluconato cálcico (90 mg de calcio elemento por 1 g) o de carbonato cálcico (400 mg de calcio elemento por 1 g) para aportar 1 a 2 g de Ca elemento por día. Aunque puede ser suficiente cualquier preparado de vitamina D, los compuestos hidroxilados como el calcitriol [1,25(OH)2D3] y los análogos seudohidroxilados en posición 1, como el dihidrotaquisterol, ofrecen la ventaja de un comienzo más rápido de la acción y de una eliminación más rápida del organismo. El calcitriol es especialmente útil en la insuficiencia renal, porque no requiere transformación metabólica renal. Los Pacientes con hipoparatiroidismo suelen responder al calcitriol a dosis de 0,5 a 2 m g/día v.o. El seudohipoparatiroidismo puede controlarse a veces sólo con suplementos orales de Ca. También se observa un beneficio con el calcitriol, pero se necesitan dosis más altas, de 1 a 3 m g/día.
En cualquier caso, el tratamiento con vitamina D no será eficaz a no ser que se proporcione suficiente Ca dietético o suplementado (1 a 2 g de calcio elemento por día) y fosfato. La toxicidad por vitamina D con síntomas graves de hipocalcemia puede ser una grave complicación del tratamiento con análogos de la vitamina D. Debe vigilarse frecuentemente la concentración plasmática de Ca: semanalmente al principio y después a intervalos de 1 a 3 meses después de que los niveles de Ca se hayan estabilizado. La dosis de mantenimiento de calcitriol o dihidrotaquisterol suele reducirse con el tiempo.
En los Pacientes con insuficiencia renal hay que combinar el tratamiento de la hipocalcemia con la restricción dietética de PO4 y agentes fijadores de PO4, como el carbonato cálcico, para evitar la hiperfosfatemia y las calcificaciones metastásicas. Se ha producido acumulación de aluminio en el hueso en la osteomalacia grave, o en el cerebro, causando la demencia de la diálisis, tras el empleo intensivo de fijadores de PO4 que contienen aluminio en Pacientes sometidos a diálisis. Por consiguiente, los compuestos que contienen aluminio deben evitarse en los Pacientes con insuficiencia renal, y de un modo especial en los sometidos a diálisis prolongada. A pesar del uso de fijadores de PO4, se debe insistir siempre en una restricción dietética de PO4. La administración de vitamina D es potencialmente peligrosa en la insuficiencia renal y debe limitarse a los Pacientes con síntomas de osteomalacia no relacionados con el aluminio o con un hipoparatiroidismo secundario mientras el Ca plasmático sea Ca <11 m g/dl (<2,75 m mol/l) o a Pacientes con hipocalcemia tras una paratiroidectomía. La eficacia y la duración relativamente corta de la acción del calcitriol lo convierten en el fármaco de elección en esos Pacientes. Aunque el calcitriol oral suele administrarse junto con Ca oral para suprimir el hipoparatiroidismo secundario, los resultados han sido variables en los Pacientes con una nefropatía terminal. En esos Pacientes puede ser más útil la forma parenteral del calcitriol para prevenir el hiperparatiroidismo secundario, dado que los niveles plasmáticos más altos que se alcanzan con el 1,25(OH)2D3 inhiben directamente la liberación de PTH. Una osteomalacia simple responde a niveles tan bajos como 0,25 a 0,5 m g/d de calcitriol oral, mientras que la corrección de una hipocalcemia posparatiroidectomía puede requerir una administración prolongada de hasta 2 mg de calcitriol y ³2 g de calcio por día. La osteoporosis causada por el aluminio suele presentarse en Pacientes con nefropatía en fase terminal sometidos a diálisis que han tomado grandes cantidades de fijadores de PO4 que contienen aluminio. En estos Pacientes, la eliminación del aluminio con desferoxamina es imprescindible antes de que mejoren las lesiones óseas con el calcitriol.
El raquitismo por deficiencia de vitamina D responde a cantidades tan pequeñas como 10 mg (400 UI/d) de vitamina D2 o D3; si existe una osteomalacia manifiesta se administran 125 m g/d (5.000 UI/d) de vitamina D2 o D3 durante 6 a 12 sem y después se reducen a 10 m g/d (400 UI/d). Durante las etapas iniciales es deseable un tratamiento adicional con 2 g calcio/d. En Pacientes con raquitismo u osteomalacia debidos a falta de exposición a la luz solar, puede ser suficiente el tratamiento con aumento de exposición a la luz solar o a la lámpara ultravioleta.
El raquitismo dependiente de vitamina D tipo I responde a dosis fisiológicas de calcitriol de 0,25 a 1,0 m g/d v.o. Los Pacientes con raquitismo dependiente de vitamina D tipo II no responden a ninguna forma de vitamina D [se ha sugerido para esta entidad el término más comprensible de «resistencia hereditaria a la 1,25(OH)2D»]. El tratamiento del raquitismo dependiente de vitamina D tipo II varía en función de la gravedad de las lesiones óseas y de la hipocalcemia. En los casos más graves se precisan hasta 6 mg/kg de peso corporal o un total de 30 a 60 m g/d de calcitriol, además de hasta 3 g de calcio elemento diarios. El tratamiento con vitamina D exige la monitorización de los niveles plasmáticos de Ca; aunque puede producirse hipercalcemia, responde en general con rapidez al ajuste de la dosis de vitamina D.
Hipercalcemia
Aumento de la concentración de calcio plasmático total por encima de 10,4 m g/dl (2,60 m mol/l).
Etiología y patogenia
La hipercalcemia suele ser el resultado de una reabsorción ósea excesiva. Las principales causas de hipercalcemia se enumeran en la tabla 12-7 y se describen a continuación.

El hiperparatiroidismo primario es un trastorno generalizado debido a una secreción excesiva de hormona paratiroidea por una o más glándulas paratiroides. El hiperparatiroidismo primario es probablemente la causa más frecuente de hipercalcemia en la población general. La incidencia del hiperparatiroidismo aumenta con la edad y es mayor en las mujeres posmenopáusicas. Se presenta con gran frecuencia tres o más décadas tras la irradiación del cuello. Existen formas familiares y esporádicas. Las formas familiares debidas a adenoma paratiroideo se producen en asociación con otros tumores endocrinos (v. cap. 10). El examen histológico muestra un adenoma paratiroideo aproximadamente en el 90% de los Pacientes, aunque a veces es difícil distinguir un adenoma de la glándula normal. Aproximadamente un 7% de los casos se deben a hiperplasia de dos o más glándulas. El cáncer de paratiroides se produce raras veces en un 3% de los casos.
El síndrome de hipercalcemia hipocalciúrica familiar se transmite como un rasgo dominante autosómico. Se caracteriza por hipercalcemia persistente, a menudo desde una edad temprana, niveles elevados de PTH e hipocalciuria. Este síndrome, que se asocia con hiperplasia paratiroidea, se considera a veces un trastorno de la detección del Ca por la glándula paratiroides.
El hiperparatiroidismo secundario aparece cuando la hipocalcemia crónica, que está causada por un trastorno como la insuficiencia renal o los síndromes de malabsorción intestinal, estimulan un aumento de la secreción de PTH. Una vez que llega a establecerse el hiperparatiroidismo secundario, puede haber hipercalcemia o, menos frecuentemente, normocalcemia. La sensibilidad de la paratiroides al Ca puede estar disminuida a causa de la pronunciada hiperplasia glandular y de la elevación del punto de ajuste del Ca (es decir, la cantidad de Ca necesaria para reducir la secreción de PTH). El hiperparatiroidismo terciario es el resultado de una hipersecreción autónoma de PTH con independencia de la concentración plasmática de Ca. El hiperparatiroidismo terciario se presenta generalmente en Pacientes con una nefropatía avanzada e hiperparatiroidismo secundario de larga evolución.
La hipercalcemia en los Pacientes hospitalizados se debe muy a menudo a procesos malignos. Aunque los tumores malignos pueden causar hipercalcemia por varios mecanismos diferentes, la elevación del Ca plasmático se produce en todos ellos como consecuencia de la reabsorción ósea. Los cánceres hematológicos -con mucha frecuencia los mielomas, pero también ciertos linfomas y linfosarcomas- causan hipercalcemia mediante la elaboración de un grupo de citocinas que estimulan a los osteoclastos a reabsorber el hueso, con producción de lesiones osteolíticas y/u osteopenia difusa. Con gran frecuencia, la hipercalcemia de la malignidad es causada por tumores cancerosos sólidos con metástasis óseas. El cáncer de mama con metástasis óseas constituye >50% de los Pacientes con hipercalcemia asociada a procesos malignos. En estos Pacientes, la hipercalcemia es el resultado de la elaboración local de citocinas o prostaglandinas activadoras de los osteoclastos y/o reabsorción ósea directa por las células tumorales metastásicas.
La hipercalcemia humoral de los tumores malignos se produce con mucha frecuencia asociada a diversos carcinomas de células escamosas, carcinomas de células renales, cáncer de mama o cáncer ovárico. Muchos casos se atribuyeron al principio a una producción ectópica de PTH, puesto que, en la hipercalcemia humoral de los tumores malignos, la hipercalcemia se presenta sin metástasis óseas detectables; sin embargo, los datos más recientes han establecido que los tumores malignos no paratiroideos producen raras veces PTH. Los ensayos obtienen niveles de PTH indetectables o intensamente inhibidos en la mayoría de los Pacientes, a pesar de la presencia de hipofosfatemia, fosfaturia y niveles elevados de AMPc nefrogénico. En varios tumores asociados con hipercalcemia humoral se ha aislado un péptido afín a la PTH que se une a los receptores de PTH en el hueso y en el riñón e imita muchos de los efectos de la hormona. El péptido es de mayor tamaño que la PTH, pero se parece a la hormona nativa en su secuencia N-terminal. Aunque la prevalencia y la identidad de esos péptidos aún no se ha establecido en diversos tumores, parece ser que la principal causa de la hipercalcemia humoral de los tumores malignos es una reabsorción ósea por osteoclastos, la cual está mediada por un péptido afín a la PTH y/o por otros factores elaborados por el tumor.
Aunque las concentraciones plasmáticas de 1,25(OH)2D3 son bajas en la mayoría de los Pacientes con tumores sólidos, los niveles plasmáticos aumentados de 1,25(OH)2D3 pueden causar rara vez hipercalcemia en asociación con un linfoma o un leiomioblastoma.
La vitamina D exógena a dosis farmacológicas produce una reabsorción excesiva de hueso y asimismo un aumento de la reabsorción intestinal de Ca e hipercalciuria (v. Toxicidad de la vitamina D, cap. 3).
La sarcoidosis se asocia con hipercalcemia hasta en un 20% de los Pacientes y con hipercalciuria hasta en un 40%. La hipercalcemia y la hipercalciuria se han descrito también en otras enfermedades granulomatosas, como tbc, lepra, beriliosis, histoplasmosis y coccidioidomicosis. En la sarcoidosis, la hipercalcemia y la hipercalciuria parecen deberse a una conversión no regulada de 25(OH)D3 a 1,25(OH)2D3 presumiblemente a causa de la expresión de la enzima 1-a-hidroxilasa en las células mononucleares de los granulomas sarcoideos. Análogamente, se han descrito niveles plasmáticos elevados de 1,25(OH)2D3 en Pacientes hipercalcémicos con tbc, silicosis y linfoma. Otros mecanismos serían responsables de la hipercalcemia en algunos casos, puesto que se han descrito niveles de 1,25(OH)2D3 disminuidos en algunos Pacientes con hipercalcemia asociada con lepra, linfoma de células T o leucemia.
La inmovilización, particularmente si es completa, y el reposo prolongado en cama, pueden producir una hipercalcemia debida a una aceleración de la reabsorción ósea. La hipercalcemia aparece en el curso de días a semanas desde el comienzo del reposo en cama en Pacientes sometidos a yesos ortopédicos o a tracción, y en los que tienen lesiones vertebrales o trastornos neurológicos. La regresión de la hipercalcemia tiene lugar inmediatamente al reemprender el soporte de peso.
La hipercalcemia idiopática de la infancia se debe a un grupo de trastornos genéticos infrecuentes; todos ellos se asocian con aumento de la reabsorción intestinal de Ca y pueden ser el resultado de toxicidad por vitamina D o aumento de sensibilidad a la vitamina D. Un enriquecimiento reducido de vitamina D de la leche puede conducir a una disminución de este trastorno, pero todavía se presentan casos asociados con la intoxicación materna por la vitamina, hiperparatiroidismo neonatal e hipocalciuria hipercalcémica familiar.
En el síndrome de la leche y los alcalinos se ingieren cantidades excesivas de Ca y álcalis absorbibles, generalmente durante el tratamiento de la úlcera péptica, originándose hipercalcemia, insuficiencia renal y alcalosis metabólica. La disponibilidad del tratamiento con bloqueantes H2 para la enfermedad ulcerosa péptica ha reducido en gran parte la incidencia de este síndrome. Cuando se sospecha un síndrome de la leche y los alcalinos, está justificada una nueva evaluación para descartar otras causas de hipercalcemia y síntomas de enfermedad ulcerosa péptica, como en el hiperparatiroidismo que se presenta con un tumor del tipo Zollinger-Ellison (v. cap. 10). La ingestión crónica de dosis altas de carbonato cálcico, generalmente para prevenir la osteoporosis, especialmente si está asociada a un tratamiento diurético con tiazidas, se ha descrito que causa una grave hipercalcemia en algunos Pacientes.
Síntomas, signos y diagnóstico
En la hipercalcemia leve, muchos Pacientes están asintomáticos. El trastorno se descubre con frecuencia accidentalmente en un análisis de rutina de laboratorio. Las manifestaciones clínicas de la hipercalcemia son estreñimiento, anorexia, náuseas y vómitos, dolor abdominal e íleo. El deterioro del mecanismo de concentración renal conduce a poliuria, nicturia y polidipsia. La elevación del Ca plasmático >12 m g/dl (>3,00 m mol/l) está asociada con labilidad emocional, confusión, delirio, psicosis, estupor y coma. La afectación neuromuscular puede causar una importante debilidad de los músculos esqueléticos. Las convulsiones son raras. La hipercalciuria con nefrolitiasis es frecuente. Con menor frecuencia, la hipercalcemia prolongada o grave puede producir insuficiencia renal aguda reversible o lesión renal irreversible debida a nefrocalcinosis (precipitación de sales de Ca en el interior del parénquima renal). La úlcera péptica y la pancreatitis también pueden asociarse con el hiperparatiroidismo, pero la relación entre estas patologías y la hipercalcemia sigue siendo oscura.
La hipercalcemia grave se asocia con acortamiento del intervalo QTC en el ECG y pueden presentarse arritmias cardíacas, especialmente en Pacientes que toman digital. Una hipercalcemia que supere 18 m g/dl (4,50 m mol/l) puede conducir a shock, insuficiencia renal y muerte.
El hiperparatiroidismo grave o de duración prolongada conduce a veces a lesiones óseas de osteítis fibrosa quística, especialmente en Pacientes en diálisis prolongada con hiperparatiroidismo secundario. En este trastorno, el aumento de la actividad osteoclástica debida a la sobreestimulación por la PTH causa rarefacción del hueso con degeneración fibrosa y formación de quistes y nódulos fibrosos. La exploración radiográfica muestra típicamente quistes óseos con aspecto de «sal y pimienta» del cráneo y una reabsorción subperióstica del hueso en las falanges y en la parte distal de las clavículas.
El hiperparatiroidismo primario suele caracterizarse por hipercalcemia, hipofosfatemia y reabsorción ósea excesiva. Aunque la hipercalcemia asintomática es la presentación más frecuente, la nefrolitiasis también es común, especialmente cuando la hipercalciuria es de larga duración.
En el síndrome de hipercalcemia hipocalciúrica familiar, la hipercalcemia suele ser asintomática, la función renal está bien conservada y la nefrolitiasis es infrecuente. Sin embargo, se presenta a veces una pancreatitis grave. Los síntomas pueden estar también presentes en lactantes gravemente enfermos familiares de personas afectadas.
La causa subyacente a la hipercalcemia suele ser manifiesta a partir de la historia y los hallazgos clínicos asociados. La evidencia radiográfica de la enfermedad ósea puede sugerir el diagnóstico por la demostración de lesiones osteolíticas u osteoblásticas o de las lesiones características del hiperparatiroidismo.
Datos de laboratorio
En el hiperparatiroidismo, el Ca plasmático es rara vez >12 m g/dl (3,00 m mol/l), pero el Ca plasmático ionizado está casi siempre elevado. Un nivel de PO4 plasmático bajo sugiere alguna forma de hiperparatiroidismo, especialmente cuando se asocia con un aclaramiento elevado de PO4 (es decir, una reabsorción tubular de PO4 deprimida) e hipercloremia leve (con o sin acidosis).
En presencia de insuficiencia renal es difícil distinguir el hiperparatiroidismo primario del secundario. Un Ca plasmático alto y un PO4 plasmático normal sugieren un hiperparatiroidismo primario, especialmente en el Paciente no dializado. La presencia de un PO4 elevado sugiere hiperparatiroidismo secundario.
Cuando el hiperparatiroidismo conduce a un aumento del recambio óseo, la fosfatasa alcalina suele estar aumentada. El nivel de PTH intacta suele estar elevado, pero se interpreta mejor conjuntamente con la concentración de Ca ionizado. En los Pacientes con hiperparatiroidismo la PTH está elevada inadecuadamente (es decir, en ausencia de hipocalcemia). La PTH está inhibida en Pacientes con la mayoría de las demás causas de hipercalcemia (v. más atrás comentario de la hormona paratiroidea en Metabolismo del calcio).
La hipercalcemia humoral de los tumores malignos puede asociarse con disminución de PO4, alcalosis metabólica e hipoalbuminemia. Sin embargo, el diagnóstico se hace por la presencia del péptido afín a la PTH. Un Ca plasmático >12 m g/dl (3,00 m mol/l) suele indicar la presencia de tumores u otra causa de hipercalcemia al lado del hiperparatiroidismo. Se sospecha un mieloma ante el síndrome de anemia, azoemia e hipercalcemia. Este diagnóstico se confirma mediante examen de la médula ósea o por la presencia de una gammapatía monoclonal indicada por la presencia de una sola especie de inmunoglobulinas o de cadenas ligeras libres en el plasma o la orina mediante inmunoelectroforesis.
La hipercalcemia hipocalciúrica familiar se distingue del hiperparatiroidismo primario por la edad temprana de aparición, la incidencia frecuente de hipermagnesemia y la presencia de hipercalcemia sin hipercalciuria en otros miembros de la familia. La fracción de excreción de Ca (proporción de aclaramiento de Ca respecto al aclaramiento de creatinina) es baja (<1%) en los Pacientes con hipercalcemia hipocalciúrica familiar; está casi siempre elevada (1 a 4%) en el hiperparatiroidismo primario. La hipercalciuria se encuentra en la mayoría de los demás trastornos que causan hipercalcemia, excepto en el síndrome de la leche y los alcalinos, el tratamiento con tiazidas y la insuficiencia renal. La PTH intacta puede estar elevada o normal en la hipercalcemia hipocalciúrica familiar, tal vez como reflejo de la alteración de la regulación por retroalimentación de las glándulas paratiroideas.
La hipercalcemia idiopática de la infancia se reconoce por la combinación de niveles de PTH inhibidos, hipercalciuria y, en algunos Pacientes gravemente afectados, anomalías somáticas del síndrome de Williams (p. ej., estenosis aórtica supravalvular, retraso mental y facies de duendecillo).
El síndrome de la leche y los alcalinos se identifica por la historia y por una combinación de hipercalcemia, alcalosis metabólica y, a veces, azoemia con hipocalciuria. Cuando se interrumpe la ingestión de Ca y álcalis, el nivel plasmático de Ca vuelve rápidamente a la normalidad, aunque la insuficiencia renal puede persistir si existe nefrocalcinosis.
En otras causas endocrinas de hipercalcemia, como en la tirotoxicosis o la enfermedad de Addison, los hallazgos de laboratorio típicos del trastorno subyacente ayudan a establecer el diagnóstico (v. caps. 8 y 9).
La PTH circulante suele elevarse en los Pacientes con hiperparatiroidismo y está suprimida en los Pacientes con toxicidad por vitamina D, en el síndrome de la leche y los alcalinos y en la sarcoidosis. La mayor parte de los Pacientes con hipercalcemia humoral por tumores malignos tienen una PTH suprimida o indetectable. Dado que muchos de estos Pacientes tendrán fosfaturia, hipofosfatemia y una elevada excreción de AMPc urinario, el hallazgo adicional de una PTH suprimida servirá para distinguir a estos Pacientes de los que tienen hiperparatiroidismo primario. En la hipercalcemia asociada con sarcoidosis, otras enfermedades granulomatosas y algunos linfomas, los niveles de 1,25(OH)2D3 pueden estar elevados.
Tratamiento
El tratamiento de la hipercalcemia depende de la presencia de síntomas, de la gravedad de elevación del Ca y de la causa subyacente. Cuando los síntomas son leves y el Ca plasmático es <11,5 m g/dl (<2,88 m mol/l), la corrección del trastorno subyacente suele bastar. Cuando el Ca plasmático es superior a 15 m g/dl (3,75 m mol/l), o cuando existen signos clínicos graves de hipercalcemia, es imprescindible un tratamiento directo para reducir el Ca plasmático.
Solución salina y furosemida: El pilar principal del tratamiento en los Pacientes con una función renal relativamente normal es aumentar la excreción renal de Ca mediante expansión del volumen extracelular con solución salina i.v. y la administración de furosemida. El objetivo es obtener un volumen de orina de 3 l/d como mínimo. Los Pacientes hipercalcémicos suelen tener un déficit de volumen preexistente y debe reponerse antes de iniciar la diuresis con infusión de solución salina al 0,9%. Durante el tratamiento diurético para la hipercalcemia, se debe permitir a los Pacientes un acceso libre al agua. El volumen eliminado por orina debe reemplazarse con solución salina al 0,9% i.v. con KCl suficiente para prevenir la hipopotasemia. Durante el tratamiento deben monitorizarse cuidadosamente la ingesta de líquido, la diuresis y los electrólitos plasmáticos.
Aunque no existe ningún método completamente satisfactorio para corregir la hipercalcemia grave en los Pacientes con insuficiencia renal, tal vez el método más seguro y fiable sea la hemodiálisis inmediata con líquido de diálisis de baja concentración de Ca o exento de Ca.
Fosfatos i.v.: Un enfoque de tratamiento más peligroso es la administración i.v. de fosfato disódico y monopotásico. No se debe administrar más de 0,5 a 1,0 g por vía i.v. en 24 h; generalmente serán suficientes 1 o 2 dosis a lo largo de 2 d para reducir el Ca plasmático durante 10 a 15 d. La disminución del Ca plasmático con este tipo de tratamiento se asocia con calcificación de tejidos blandos y puede presentarse una insuficiencia renal aguda. El PO4 i.v. sólo debe utilizarse cuando la hipercalcemia constituye una amenaza para la vida y es refractaria a otros métodos y cuando no se puede realizar la hemodiálisis inmediata. La infusión i.v. de sulfato sódico es incluso más peligrosa y menos eficaz que la infusión de fosfato y no debe utilizarse.
Mitramicina: La mitramicina (Plicamycin) en dosis de 25 mg/kg i.v. en 50 ml de solución de dextrosa al 5% a lo largo de 3 a 6 h es extraordinariamente eficaz en Pacientes con metástasis esqueléticas o hipercalcemia humoral de los tumores malignos. La mitramicina reduce el Ca plasmático durante 12 a 36 h. Sin embargo, su utilidad es limitada en el tratamiento prolongado, dada la toxicidad del fármaco, la duración variable de la acción (de varios días a 3 sem) y porque produce un efecto de rebote de la hipercalcemia que puede ser rápido y grave. La mitramicina puede causar trombocitopenia, defecto cualitativo de las plaquetas (diátesis hemorrágica con un recuento de plaquetas normal), hepatotoxicidad y lesión renal. Su potencial de toxicidad puede reducirse manteniendo un intervalo entre las dosis de 72 h como mínimo. En Pacientes con anomalías de la hematopoyesis o de la función hepática o renal son preferibles otros fármacos. Si es preciso utilizar la mitramicina en esa clase de Pacientes, puede ser aconsejable reducir la dosis a 12,5 mg/kg. La mitramicina puede reemplazarse por nitrato de galio o bien por difosfonatos cuando la eficacia y la seguridad a largo plazo de estos fármacos más recientes esté firmemente establecida.
Calcitonina (tirocalcitonina): La calcitonina es una hormona peptídica de acción rápida secretada por las células parafoliculares del tiroides (células C), en respuesta a la hipercalcemia. Parece ser que la calcitonina reduce el Ca plasmático mediante inhibición de la actividad osteoclástica y con ello la liberación de Ca procedente del hueso. Se dispone de un preparado comercial de calcitonina de salmón que resulta muy útil en el tratamiento de la enfermedad de Paget. Se ha sugerido que la administración de calcitonina de salmón (4 a 8 UI/kg s.c. cada 12 h) y prednisona (30 a 60 m g/día v.o. fraccionada en tres dosis) puede controlar la grave hipercalcemia asociada con los procesos malignos, incluso en Pacientes con nefropatía en quienes no es aconsejable el tratamiento primario con solución salina i.v. Su utilidad en el tratamiento de la hipercalcemia asociada con los procesos malignos se ve limitada por la breve duración de la acción y la falta de respuesta hasta en un 25% de los Pacientes. Sin embargo, la combinación de calcitonina de salmón y prednisona puede controlar el Ca plasmático incluso durante varios meses en algunos Pacientes con tumores malignos. Si la calcitonina deja de actuar, puede interrumpirse su administración durante 2 d (al mismo tiempo que se continúa con prednisona) y reanudar después el tratamiento.
Corticosteroides: La adición de prednisona, 20 a 40 m g/d v.o. controlará eficazmente la hipercalcemia en la mayoría de los Pacientes con toxicidad por vitamina D, hipercalcemia idiopática de la infancia y sarcoidosis. Algunos Pacientes con mieloma, linfoma, leucemia o cáncer de mama metastásico responden con 40 a 60 m g/d de prednisona. Sin embargo, dado que la respuesta a los glucocorticoides necesita varios días y que más del 50% de los Pacientes con hipercalcemia debida a esos tumores no responden a los glucocorticoides, suele ser necesario otro tratamiento.
Nitrato de galio: Se ha descrito que el nitrato de galio reduce el Ca plasmático eficazmente en la hipercalcemia asociada a metástasis óseas, hipercalcemia humoral de los tumores malignos y cáncer de paratiroides. Parece ser que el nitrato de galio inhibe la reabsorción ósea por los osteoclastos. La duración media de la normocalcemia en el tratamiento con galio es de unas 2 sem. La infusión de nitrato de galio está indicada tras el fracaso de la solución salina y los diuréticos del asa en el control de la hipercalcemia asociada con los tumores malignos, aunque su eficacia con el uso repetido no se ha establecido y el tratamiento prolongado requiere una evaluación ulterior. Parece tener pocos efectos adversos aparte de la hipocalcemia, la hipofosfatemia y la nefrotoxicidad. El nitrato de galio puede causar insuficiencia renal aguda y no debe utilizarse en Pacientes con deterioro renal grave o simultáneamente con otros fármacos nefrotóxicos. Además, el galio debe administrarse solamente a Pacientes con un volumen intravascular normal. Deben monitorizarse con frecuencia las concentraciones plasmáticas de creatinina, Ca y PO4.
Bisfosfonatos: Los bisfosfonatos, que actúan inhibiendo la reabsorción ósea por los osteoclastos, se utilizan en la actualidad ampliamente como tratamiento de primera línea en unión con la solución salina y la furosemida en la hipercalcemia asociada a los procesos malignos. El etidronato disódico ha estado disponible en Estados Unidos durante varios años como tratamiento eficaz para controlar la reabsorción ósea en la enfermedad de Paget. Su uso en la hipercalcemia asociada a tumores malignos estuvo limitado por la toxicidad renal. El palmidronato y el clodronato parecen ser más seguros. Ambos se administran en forma de infusión i.v. y reducen las concentraciones plasmáticas de Ca a lo largo de 5 a 7 d. Los efectos secundarios son fiebres y mialgias pasajeras. A veces puede aparecer leucopenia, hipocalcemia con síntomas e hipofosfatemia.
Fosfato de cloroquina: El fosfato de cloroquina, 500 m g/d v.o., puede inhibir la síntesis de la 1,25(OH)2D3 y reducir los niveles de Ca plasmático en Pacientes con sarcoidosis. Es obligatoria la vigilancia oftalmológica de rutina durante la administración crónica de cloroquina, puesto que puede presentarse lesión de la retina relacionada con la dosis.
Tratamiento quirúrgico: En el hiperparatiroidismo, el tratamiento es quirúrgico si la enfermedad es sintomática o progresiva. El resultado de la cirugía depende de la extirPación con éxito de todo el tejido funcionante en exceso. Es preciso extirpar todas las glándulas adenomatosas. El tejido paratiroideo restante también suele extirparse, dado que es normalmente muy difícil localizar las glándulas paratiroides durante la exploración quirúrgica subsiguiente. Para evitar un hipoparatiroidismo secundario, suele reimplantarse una pequeña porción de glándula paratiroides de aspecto normal en el vientre del músculo esternocleidomastoideo o por vía subcutánea en el antebrazo. También se ha llevado a cabo a veces la crioconservación de tejido paratiroideo para hacer posible un trasplante autólogo ulterior si se presenta un hipoparatiroidismo persistente. Las glándulas paratiroideas anormalmente funcionantes pueden encontrarse en localizaciones inhabituales en el cuello y el mediastino, y requieren un cirujano experimentado para encontrarlas. Cuando un cirujano experto lleva a cabo la exploración de las paratiroides por primera vez, son habituales unas tasas de curación del 90% y es innecesaria la localización preoperatoria de rutina del tejido paratiroideo. En estos Pacientes, la TC de alta resolución, con biopsia guiada por TC o sin ella, y el inmunoensayo del drenaje venoso tiroideo parecen ser más sensibles y específicos que la ecografía de alta resolución, la angiografía por sustracción digital o la gammagrafía con talio 201-tecnecio 99. El tecnecio 99 sestamibi, un nuevo agente radioisotópico para estudio de las paratiroides, es más sensible y específico que otros agentes más antiguos, y se emplea cada vez más.
Las indicaciones de la cirugía en Pacientes con un hiperparatiroidismo primario leve y asintomático están pendientes de aclaración. Los datos indican que los Pacientes con hiperparatiroidismo primario asintomático pueden tratarse de forma conservadora en ausencia de hipercalcemia progresiva u otras complicaciones; sin embargo, la preocuPación respecto a una enfermedad ósea subclínica, la hipertensión y la longevidad subsiste.
Cuando el hiperparatiroidismo es leve no se necesitan precauciones postoperatorias especiales. El nivel plasmático de Ca desciende a un valor inmediatamente inferior al normal en 24 a 48 h tras la operación. En los Pacientes con osteítis fibrosa quística grave, la carga con 10 a 20 g de calcio elemento en los días anteriores a la cirugía puede atenuar la hipocalcemia sintomática prolongada que puede presentarse tras la operación. Aun cuando reciban Ca en el período preoperatorio, estos Pacientes necesitan grandes dosis de Ca y vitamina D hasta que se reponga el calcio óseo (v. más atrás, Hipocalcemia).
En el síndrome de hipercalcemia hipocalciúrica familiar, a pesar de que la hiperplasia se encuentra de manera consistente, la respuesta a la paratiroidectomía subtotal no es satisfactoria. Dado que las manifestaciones clínicas son evidentes en la hipercalcemia hipocalciúrica familiar, es poco frecuente que se precise otro tratamiento específico que no sea el de la necesidad ocasional de medidas para reducir el calcio descritas arriba. No obstante, si se diesen la pancreatitis o el hiperparatiroidismo primario neonatal, la intervención de elección sería la paratiroidectomía total.
METABOLISMO DEL FOSFATO
El fósforo es uno de los elementos más abundantes en el cuerpo humano. La mayor parte del fósforo en el cuerpo constituye un complejo con el oxígeno en forma de fosfato (PO4). Alrededor del 85% de los 500 a 700 g de PO4 que presenta aproximadamente el organismo están contenidos en el hueso, donde es un importante constituyente de la hidroxiapatita cristalina. En los tejidos blandos el PO4 se encuentra sobre todo en el compartimiento intracelular. Es parte integrante de varios compuestos orgánicos, como los ácidos nucleicos y los fosfolípidos de la membrana celular. El PO4 también está íntimamente implicado en el metabolismo energético aerobio y anaerobio. El 2,3-difosfoglicerato (2,3-DPG) de los eritrocitos representa un papel crucial en el suministro de O2 a los tejidos. El PO4 inorgánico es un importante anión intracelular, pero también está presente en el plasma. La concentración de PO4 inorgánico en el plasma normal oscila entre 2,5 y 4,5 m g/dl (0,81 a 1,45 m mol/l). El PO4 es hasta un 50% más alto en los lactantes y un 30% superior en los niños, debido posiblemente a sus mayores requerimientos de fosfato para el crecimiento.
La dieta estadounidense típica contiene alrededor de 800 a 1.500 mg de PO4. Esta cantidad se presenta en las heces con cifras variables en función del número de compuestos que fijan PO4 (principalmente Ca) en la dieta. Como en el caso del Ca, la absorción gastrointestinal del PO4 también es facilitada por la vitamina D. La excreción renal de PO4 equivale aproximadamente a la absorción GI para mantener el equilibrio neto del PO4. La depleción de PO4 puede presentarse en diversos estados patológicos y conduce a la conservación del PO4 por los riñones. El PO4 óseo funciona como un depósito, el cual puede amortiguar los cambios en el PO4 plasmático e intracelular.
TRASTORNOS DEL METABOLISMO DEL FOSFATO
Hipofosfatemia
Disminución de la concentración de fosfato plasmático por debajo de 2,5 m g/dl (0,81 m mol/l).
Incidencia, etiología y patogenia
Se observa hipofosfatemia en un 2% de los Pacientes hospitalizados, pero es más prevalente en ciertas poblaciones, por ejemplo, en los alcohólicos, en quienes se observa hasta en un 10% de los Pacientes hospitalizados. Las situaciones clínicas comunes de hipofosfatemia grave son la fase de recuperación de la cetoacidosis diabética, el alcoholismo agudo y las quemaduras graves. También puede presentarse hipofosfatemia en los Pacientes que reciben nutrición parenteral total y en la alcalosis respiratoria crónica grave.
La hipofosfatemia tiene numerosas causas, pero una hipofosfatemia clínicamente importante ocurre en relativamente pocas situaciones. La hipofosfatemia crónica se produce muy a menudo por una disminución de la reabsorción renal de PO4 y no se asocia con depleción del PO4 intracelular. Entre las causas están el hiperparatiroidismo, otras alteraciones hormonales, como el síndrome de Cushing y el hipotiroidismo, trastornos electrolíticos, como la hipomagnesemia y la hipopotasemia, la intoxicación por teofilina y la administración crónica de diuréticos. La hipofosfatemia crónica grave suele ser consecuencia de un balance de PO4 negativo prolongado. Las causas son la inanición o la malabsorción crónicas, especialmente si se combinan con vómitos y diarreas copiosos o ingestión crónica de grandes cantidades de aluminio fijador de PO4, habitualmente en forma de antiácidos. Estos últimos son especialmente propensos a producir depleción de PO4 cuando se combinan con una disminución de la ingesta dietética y pérdidas de PO4 por diálisis en Pacientes con una nefropatía en fase terminal.
La hipofosfatemia aguda con un fósforo plasmático <1 m g/dl (0,32 m mol/l) es causada muy a menudo por desplazamientos transcelulares de PO4, que coinciden frecuentemente con hipofosfatemia crónica y depleción de PO4.
Síntomas, signos y diagnóstico
Aunque la hipofosfatemia suele ser asintomática, en la depleción crónica grave puede aparecer anorexia, debilidad muscular y osteomalacia. Puede haber graves alteraciones neuromusculares, incluso encefalopatía progresiva, coma y muerte. La debilidad muscular de la hipofosfatemia grave puede ir acompañada de rabdomiólisis, especialmente en el alcoholismo agudo. Las alteraciones hematológicas de la hipofosfatemia grave incluyen anemia hemolítica, disminución de la liberación del O2 de la hemoglobina y deterioro de la función de los leucocitos y las plaquetas.
Tratamiento
El tratamiento es empírico y está dictado por la causa subyacente y la gravedad de la hipofosfatemia. En la depleción crónica de PO4 leve a moderada se puede usar fosfato sódico o potásico oral, pero suelen tolerarse mal debido a la diarrea. La ingestión de 1 litro de leche pobre en grasa o descremada proporcionará 1 g de PO4 y es más aceptable. Si es posible, debe intentarse la eliminación de la causa de la hipofosfatemia, como la interrupción de los antiácidos fijadores de PO4 y de los diuréticos, o la corrección de la hipomagnesemia.
La reposición oral de PO4 suele bastar en los Pacientes asintomáticos, aun cuando la concentración plasmática sea tan baja como 1,5 a 2 m g/dl (0,48 a 0,65 m mol/l). El fosfato oral puede administrarse en dosis de hasta 3 g/d en tabletas que contienen fosfato sódico o potásico. Sin embargo, se debe administrar PO4 parenteral cuando el PO4 plasmático cae por debajo de 0,5 mEq/l (0,16 m mol/l); cuando existe rabdomiólisis, hemólisis o síntomas del SNC, o cuando la reposición oral no es factible debido a una enfermedad subyacente. En estos Pacientes, la administración i.v. de fosfato potásico (en forma de mezcla tamponada de K2HPO4 y KH2PO4) es relativamente segura siempre que la función renal esté bien conservada. La dosis parenteral usual es 2 mg/kg administrados por vía i.v. a lo largo de 6 h. Los alcohólicos pueden necesitar ³1 g/d en el curso de la nutrición parenteral. En la cetoacidosis diabética pueden necesitarse hasta 3 g de PO4 o más en las primeras 24 h; el suplemento de PO4 se suspende al reanudar la ingestión oral. En todo caso, pero particularmente cuando se administra el PO4 por vía i.v. o a Pacientes con deterioro de la función renal, los niveles plasmáticos de Ca y PO4 deben monitorizarse durante el tratamiento. En la mayoría de los casos no se deben administrar más de 7,0 mg/kg (unos 500 mg para un adulto de 70 kg) a lo largo de 6 h. La hipocalcemia, la hiperfosfatemia, la calcificación metastásica y la hiperpotasemia pueden evitarse con una monitorización estricta y evitando tasas más rapidas de administración de PO4. En Pacientes con deterioro de la función renal deben utilizarse generalmente preparados de fosfato sódico (y no los de fosfato potásico).
Hiperfosfatemia
Aumento de la concentración de fosfato plasmático por encima de 4,5 m g/dl (1,46 m mol/l).
Incidencia, etiología y patogenia
La hiperfosfatemia se produce generalmente por una disminución de la excreción renal de PO4. Una insuficiencia renal avanzada (TFG <20 ml/min) conduce a una reducción de la excreción suficiente para causar un aumento del PO4 plasmático. Los defectos en la excreción renal del PO4 en ausencia de insuficiencia renal también se presentan en el seudohipoparatiroidismo y en el hipoparatiroidismo. También se puede observar hiperfosfatemia en una administración oral excesiva de PO4 y a veces con el uso exagerado de enemas que contienen fosfato.
La hiperfosfatemia se produce también a veces como consecuencia del desplazamiento transcelular de PO4 hacia el espacio extracelular. Esto sucede con gran frecuencia en la cetoacidosis diabética (a pesar de la depleción de PO4 corporal total), en las lesiones por aplastamiento y en la rabdomiólisis no traumática, así como en las infecciones sistémicas y en el síndrome de lisis tumoral. La hiperfosfatemia representa también un papel crítico en la aparición de hiperparatiroidismo secundario y en la osteodistrofia renal en Pacientes sometidos a diálisis crónica.
Síntomas, signos y diagnóstico
La mayoría de los Pacientes con hiperfosfatemia están asintomáticos, aunque si existe una hipocalcemia concomitante puede haber síntomas de ésta, incluso tetania. Las calcificaciones de los tejidos blandos son comunes en los Pacientes con insuficiencia renal crónica, especialmente si el producto Ca x PO4 en el plasma pasa de 70 (en mEq/l) de forma crónica.
Tratamiento
El pilar principal del tratamiento de la hiperfosfatemia en Pacientes con insuficiencia renal crónica es la reducción de la ingesta de PO4. Ésta suele llevarse a cabo evitando los alimentos que contienen cantidades altas de PO4 y con el uso de antiácidos fijadores de PO4 tomados con las comidas. Con ese fin, hace tiempo se prescribían grandes cantidades de antiácidos que contenían aluminio a los Pacientes en diálisis para tomarlos con las comidas. Como consecuencia de los peligros de la osteomalacia relacionada con el aluminio, el carbonato cálcico ha sustituido a otras sales como fijador de PO4 de primera elección.
METABOLISMO DEL MAGNESIO
El magnesio (Mg) es el cuarto catión más abundante en el organismo. Un adulto de 70 kg tiene unos 2.000 mEq de Mg. Alrededor del 50% está secuestrado en el hueso y no es fácilmente intercambiable con otros compartimientos. El LEC contiene en torno al 1% del Mg total corporal. El resto reside en el compartimiento intracelular. La concentración normal del Mg en el plasma oscila entre 1,4 y 2,1 mEq/l (0,70 a 1,05 m mol/l).
El mantenimiento de la concentración plasmática de Mg es en gran medida una función de la ingesta dietética y de una conservación renal e intestinal sumamente eficiente. A los 7 d de iniciar una dieta deficiente en Mg, la excreción renal y fecal de Mg descienden aproximadamente a 1 mEq/24 h (0,5 mmol/24 h).
Alrededor del 70% del Mg plasmático es ultrafiltrado por el riñón; el resto está unido a las proteínas. Como en el caso del Ca, la unión del Mg a las proteínas depende del pH. La concentración plasmática de Mg y el contenido de Mg corporal total o el intracelular no están relacionados estrictamente. No obstante, una hipomagnesemia plasmática grave puede reflejar una disminución de los depósitos corporales de Mg.
Una amplia diversidad de las enzimas son activadas por el Mg o dependen de él. El Mg se necesita para todos los procesos enzimáticos que involucran al ATP y también lo precisan muchas de las enzimas implicadas en el metabolismo de los ácidos nucleicos. El Mg se necesita para la actividad del cofactor pirofosfato de tiamina y al parecer estabiliza la estructura de macromoléculas como el ADN y el ARN. El Mg está también íntimamente relacionado con el metabolismo del Ca y el K, pero en una forma insuficientemente conocida.
TRASTORNOS DEL METABOLISMO DEL MAGNESIO
Hipomagnesemia
Concentración plasmática de magnesio inferior a 1,4 mEq/l (0,70 m mol/l).
Una hipomagnesemia grave suele considerarse equivalente a una depleción de Mg. Sin embargo, la concentración plasmática de Mg, incluso si se determina el ion Mg libre, puede no ser un reflejo del estado de los depósitos de Mg intracelulares u óseos.
Los trastornos asociados con la deficiencia de Mg son complicados y suelen acompañarse de múltiples alteraciones metabólicas y nutricionales.
Etiología y patogenia
La depleción de Mg suele ser consecuencia de una ingesta insuficiente así como de un deterioro de la absorción renal o intestinal. Se ha descrito en asociación con la alimentación parenteral prolongada, generalmente combinada con la pérdida de líquidos corporales por aspiración gástrica o diarreas; en la lactancia (la cual aumenta el requerimiento de Mg); en situaciones de conservación renal anormal del Mg, como la hipersecreción de aldosterona, ADH u hormona tiroidea, en la hipercalcemia, en la acidosis diabética y en el tratamiento con cisplatino o con diuréticos.
La deficiencia de Mg clínicamente importante se asocia con mayor frecuencia a: 1) síndromes de malabsorción de cualquier etiología, en los cuales el aumento de Mg en las heces es probablemente proporcional al nivel de esteatorrea, más que a una deficiencia de lugares intestinales absortivos propiamente dichos; 2) malnutrición proteico-calórica (p. ej., en el kwashiorkor); 3) enfermedad paratiroidea, en la cual se produce hipomagnesemia tras la extirPación de un tumor paratiroideo, en especial si existe una osteítis fibrosa grave (se supone que el Mg transferido al hueso en rápida mineralización y la deficiencia de Mg pueden explicar la resistencia de la hipocalcemia a la corrección con vitamina D en algunos Pacientes con hipoparatiroidismo); 4) alcoholismo crónico, en el cual la hipomagnesemia se debe probablemente tanto a la ingesta insuficiente como a la excesiva excreción renal, y 5) diarrea crónica.
Síntomas y signos
Partiendo de la depleción experimental de Mg en voluntarios humanos, las manifestaciones clínicas de la deficiencia de Mg son anorexia, náuseas, vómitos, letargia, debilidad, alteración de la personalidad, tetania (p. ej., signos de Trousseau o de Chvostek positivos o espasmo carpopedal espontáneo) y temblores y fasciculaciones musculares. Los signos neurológicos, en especial la tetania, se correlacionan con el desarrollo concomitante de hipocalcemia e hipopotasemia. Se encuentran potenciales miopáticos en la electromiografía, pero también son compatibles con la hipocalcemia o la hipopotasemia. Aunque no se observa experimentalmente, es probable que la hipomagnesemia grave pueda producir convulsiones tónico-clónicas generalizadas, especialmente en niños.
Datos de laboratorio
La hipomagnesemia suele estar presente cuando la depleción de Mg es grave. La hipocalcemia y la hipocalciuria son frecuentes en Pacientes con esteatorrea, alcoholismo u otras causas de deficiencia de Mg. Puede haber hipopotasemia con aumento de excreción urinaria de K y alcalosis metabólica. Así, una hipocalcemia y una hipopotasemia inexplicadas deben sugerir la posibilidad de una depleción de Mg.
Tratamiento
El tratamiento con sales de Mg (sulfato o cloruro) está indicado cuando la deficiencia de Mg produce síntomas o está asociada con una hipomagnesemia persistente intensa <1 mEq/l (<0,5 m mol/l). En estos casos es posible un déficit cercano a 12 a 24 mg/kg. En Pacientes con una función renal intacta se debe administrar aproximadamente el doble del déficit calculado, puesto que en torno al 50% del Mg administrado se excretará en la orina. Se administra generalmente la mitad de la dosis en las primeras 24 h, y el resto a lo largo de los 4 d siguientes. La administración parenteral se reserva para los Pacientes que tienen una hipomagnesemia grave sintomática o que no pueden tolerar fármacos orales. Cuando es preciso reponer el Mg por vía parenteral, se dispone de una solución de sulfato de magnesio (MgSO4) al 10% (1 g/10 ml) para uso i.v., y de una solución al 50% (1 g/2 ml) para uso i.m. Durante el tratamiento con Mg debe monitorizarse frecuentemente el nivel de Mg plasmático, en especial cuando el Mg se administra por vía parenteral o a Pacientes con insuficiencia renal. El tratamiento se continúa hasta alcanzar un nivel de Mg plasmático normal.
En una hipomagnesemia grave con síntomas (p. ej., convulsiones generalizadas, Mg <1 mEq/l [0,5 m mol/l]), se pueden administrar de 2 a 4 g de MgSO4 por vía i.v. a lo largo de 5 a 10 min. Si las convulsiones persisten, la dosis puede repetirse hasta un total de 10 g a lo largo de las 6 h siguientes. Si las convulsiones cesan, se pueden infundir 10 g en 1 litro de solución de dextrosa al 5% en 24 h, seguidos de hasta 2,5 g cada 12 h para reponer el déficit en los depósitos de Mg totales y prevenir nuevas caídas del Mg plasmático. Cuando el Mg plasmático es <1 mEq/l (<0,5 m mol/l), pero los síntomas son menos graves, el MgSO4 se puede administrar i.v. en solución de dextrosa al 5% a una tasa de 1 g/h en forma de infusión lenta hasta unas 10 h. En los casos de hipomagnesemia menos intensos, la repleción gradual puede lograrse con la administración de dosis parenterales menores a lo largo de 3 a 5 d hasta que el nivel de Mg plasmático sea normal (v. también el uso del MgSO4 en Preeclampsia y eclampsia, cap. 252). Los Pacientes hipocalcémicos que también tienen una depleción de Mg con hipomagnesemia resultante, requieren generalmente una repleción de Mg además de la administración de Ca.
Hipermagnesemia
Concentración de magnesio plasmático superior a 2,1 mEq/l (1,05 m mol/l).
La hipermagnesemia con síntomas es bastante infrecuente, pero cuando se presenta suele hacerlo en Pacientes con insuficiencia renal tras la ingestión de fármacos que contienen Mg, como los antiácidos o los purgantes.
A las concentraciones plasmáticas de Mg de 5 a 10 mEq/l (2,5 a 5 m mol/l), el ECG muestra prolongación del intervalo PR, ensanchamiento del complejo QRS y aumento de amplitud de la onda T. Los reflejos tendinosos profundos desaparecen cuando el nivel plasmático de Mg se acerca a los 10 mEq/l (5,0 m mol/l); con una hipermagnesemia creciente aparecen hipotensión, depresión respiratoria y narcosis. Puede producirse parada cardíaca cuando los niveles sanguíneos de Mg exceden los 12 a 15 mEq/l (6,0 a 7,5 m mol/l).
Tratamiento
El tratamiento de la toxicidad grave por Mg consiste en el soporte circulatorio y respiratorio, con administración i.v. de 10 a 20 ml de gluconato cálcico al 10%. Este último puede contrarrestar muchos de los cambios inducidos por el Mg, incluso la depresión respiratoria. La administración i.v. de furosemida puede aumentar la excreción de Mg si la función renal es suficiente y se mantiene el estado de la volemia. La hemodiálisis puede ser valiosa en la hipermagnesemia grave, puesto que una fracción relativamente grande del Mg sanguíneo (alrededor del 70%) es ultrafiltrable. Si la hipermagnesemia se asocia con compromiso hemodinámico, y la hemodiálisis no es práctica, queda la opción de la diálisis peritoneal.
METABOLISMO ACIDOBÁSICO
La concentración sanguínea del ion hidrógeno (H+) se mantiene dentro de límites muy estrechos. La concentración de H+ en el plasma arterial oscila entre 37 y 43 nmol/l (37 x 10-6 a 43 x 10-6 mEq/l). El mantenimiento de H+ en tasas tan bajas es esencial para la función celular normal, a causa de la gran reactividad entre el H+ y otros compuestos, especialmente proteínas. El pH (logaritmo negativo de la concentración de H+) es una medida mucho menos incómoda de las concentraciones fisiológicas de H+ y se utiliza ampliamente en medicina clínica. El pH de la sangre arterial normal oscila entre 7,37 y 7,43.
La función pulmonar y la función renal mantienen el pH sanguíneo dentro de ese intervalo. En respuesta a las alteraciones del equilibrio acidobásico aparecen pronto cambios respiratorios en la ventilación por minuto y el pH sanguíneo se altera con rapidez por el cambio de la concentración de ácido carbónico a través de los cambios de la PCO2 en la sangre. Los riñones modifican la excreción renal de equivalentes de ácido o de base y alteran en último término la concentración plasmática de HCO3- para modificar el pH sanguíneo. Las adaptaciones renales a los cambios en el equilibrio acidobásico se producen a lo largo de varios días, mientras que los cambios impulsados por la respiración tienen lugar generalmente en minutos a horas. Tanto la función pulmonar como la función renal actúan compensando las alteraciones del equilibrio acidobásico para mantener el pH sanguíneo dentro de los márgenes normales.
Las fluctuaciones amplias de la concentración de H+ se evitan también mediante la presencia de varios tampones del pH. Estos tampones son ácidos débiles que existen en equilibrio con sus correspondientes bases conjugadas al pH fisiológico. Los tampones responden a los cambios en la [H+] mediante un desplazamiento de las concentraciones del tampón y la base conjugada correspondiente para amortiguar el cambio del pH. Los fosfatos, el amoniaco, las proteínas, incluida la hemoglobina, y el hueso poseen capacidad para tamponar el pH, pero el principal tampón del pH en la sangre, y el que es más relevante para las alteraciones acidobásicas clínicas, es el sistema bicarbonato/ácido carbónico.
La enzima anhidrasa carbónica convierte rápidamente el ácido carbónico en la sangre a CO2 y agua. La presión parcial del CO2 gaseoso (PCO2) se mide fácilmente en las muestras de sangre y es directamente proporcional al contenido sanguíneo de CO2; por consiguiente, la PCO2 se emplea para representar la concentración de ácido en el sistema. La concentración de base en el sistema se puede determinar directamente mediante la determinación de la concentración de HCO3-. Las concentraciones plasmáticas de bicarbonato y CO2 y el pH están relacionados químicamente entre sí mediante la ecuación de Henderson-Hasselbalch:
|
|
HCO3- |
|
pH = 6,1 + log |
---------------------- |
|
|
0,03 x PCO2 |
donde 6,1 es el pKa (logaritmo negativo de la constante de disociación del ácido para el ácido carbónico) y 0,03 relaciona la PCO2 con la cantidad de CO2 disuelta en el plasma. Aunque es incómoda y algo difícil de utilizar a la cabecera del Paciente, la ecuación de Henderson-Hasselbalch representa una relación funcional muy importante. Predice que la proporción de HCO3- a CO2 disuelto determina el pH sanguíneo, más que sus concentraciones actuales. Este sistema tampón es de importancia fisiológica porque ambos mecanismos para regular el pH, pulmonar y renal, funcionan mediante el ajuste de esa proporción. La PCO2 puede modificarse con rapidez con los cambios de la respiración por minuto, mientras que la concentración plasmática de [HCO3-] puede modificarse regulando su excreción por los riñones.
Las alteraciones clínicas del metabolismo acidobásico se definen clásicamente en términos del sistema tampón HCO3-/CO2. Los aumentos o disminuciones de HCO3- se denominan alcalo-sis o acidosis metabólica, respectivamente. Los aumentos o disminuciones de la PCO2 se denominan acidosis o alcalosis respiratoria, respectivamente. Las alteraciones acidobásicas simples incluyen la alteración primaria y también una compensación esperada. Por ejemplo, en la acidosis metabólica existe una caída primaria de la concentración plasmática de HCO3- y un descenso secundario de la PCO2 debido a la compensación respiratoria. La tabla 12-8 muestra los cambios primarios en las cuatro alteraciones acidobásicas simples y la compensación esperada.

Las alteraciones acidobásicas mixtas son trastornos más complejos en los cuales coexisten dos o más alteraciones primarias. Los mecanismos compensadores también existen en las alteraciones acidobásicas mixtas. Éstas se identifican generalmente cuando para una alteración primaria dada se presenta una compensación menor o mayor que la prevista. Los nomogramas hacen posible la representación gráfica simultánea del pH, el HCO3- y la PCO2 y simplifican considerablemetne la identificación de los trastornos mixtos. Es preciso dirigir el tratamiento a cada una de las alteraciones primarias. La determinación del pH, la PCO2 y el HCO3- en sangre arterial, junto con la identificación del proceso patológico subyacente, suelen bastar para resolver correctamente la mayoría de las alteraciones acidobásicas en la clínica.
Las alteraciones acidobásicas pueden afectar considerablemente al transporte de O2 y a la oxigenación de los tejidos. Los cambios agudos de la concentración de H+ influyen rápidamente sobre la curva de disociación de la oxihemoglobina (efecto Bohr); la acidemia desplaza la curva hacia la derecha (disminución de la afinidad de la Hb por el oxígeno; facilitación de la liberación de oxígeno hacia los tejidos), la alcalemia desplaza la curva hacia la izquierda (aumento de la afinidad de la Hb por el O2; disminución de la liberación de O2 hacia los tejidos). Sin embargo, cuando la acidosis o la alcalosis son crónicas, estos efectos agudos sobre la unión Hb-O2 se modifican por cambios de desarrollo más lento en las concentraciones eritrocitarias de 2,3-difosfoglicerato (2,3-DPG). Así, las elevaciones crónicas del ion H+ inhiben la formación del 2,3-DPG (con resultado de un aumento de la afinidad de la Hb por el O2), y la depresión crónica del ion H+ aumenta el 2,3-DPG (con resultado de una disminución de la afinidad de la Hb por el O2). Estos cambios agudos en el transporte del O2 y en la oxigenación tisular pueden desempeñar un papel en la producción de los síntomas del SNC de la alcalemia aguda, pero su importancia clínica en la acidosis es dudosa.
Los riñones representan un papel importante en la regulación de la concentración de HCO3- del LEC. Prácticamente la totalidad del HCO3- plasmático es filtrado por el glomérulo. Grandes cantidades de ion H+ son secretadas en la luz del túbulo proximal renal en intercambio por Na+. Por cada ion H+ secretado, sale un ion HCO3- hacia el LEC. De este modo, tiene lugar una reabsorción neta del HCO3- filtrado. Dado que el pH del líquido que abandona el túbulo proximal es aproximadamente de 6,5, la mayoría del HCO3- filtrado es reabsorbida en el túbulo proximal. En el túbulo distal, la secreción de ion H+ depende en parte de la reabsorción de Na+ mediada por la aldosterona. La reabsorción de HCO3- puede continuar en la nefrona distal a favor de un fuerte gradiente cuando el pH urinario puede disminuir en este segmento de la nefrona a valores tan bajos como 4,5 a 5,0. En toda la nefrona, el ion H+ secretado es amortiguado por los tampones urinarios, como el PO4 (ácido titulable) y el amoniaco. De esta forma, el HCO3- filtrado se reabsorbe activamente y puede generarse nuevo HCO3- para reemplazar el perdido en las reacciones amortiguadoras del organismo. Dado que el Na+ filtrado se reabsorbe, sea en asociación con Cl o mediante intercambio catiónico con el ion H+ o en menor medida con K, el Na+ total reabsorbido se aproxima a la suma del Cl reabsorbido y el ion H+ secretado. Así pues, existe una relación funcional inversa entre la reabsorción de Cl y la secreción de ion H+, que depende mucho del nivel de reabsorción de Na+ presente.
La reabsorción renal de HCO3- también está influida por los depósitos corporales de K. Existe una relación recíproca general entre el contenido de K intracelular y la secreción de ion H+. Así, la depleción de K está asociada con aumento de la secreción de ion H+ y la generación asociada de HCO3-, lo que conduce a un aumento de HCO3- en el LEC y a alcalosis metabólica. Finalmente, la reabsorción renal de HCO3- está influida por la PCO2 y el estado del balance de cloruro. El aumento de la PCO2 lleva a un aumento de la reabsorción de HCO3-. La depleción de Cl lleva a un aumento de la reabsorción de Na+ y a la generación de HCO3- por el túbulo proximal. Aunque la depleción de Cl se puede producir experimentalmente sin depleción de volumen del LEC, en situaciones clínicas la depleción de Cl es sinónima generalmente de la depleción de volumen del LEC.
TRASTORNOS DEL METABOLISMO ACIDOBÁSICO
Acidosis metabólica
Trastorno con pH arterial bajo, concentración plasmática de HCO3- reducida y, generalmente, hiperventilación alveolar compensadora que conduce a una disminución de la PCO2.
Etiología y patogenia
La acidosis metabólica se produce cuando existe un proceso que conduce a la eliminación de equivalentes de ácido en el organismo. Si la carga de ácido desborda la capacidad respiratoria, resultará una acidemia (pH arterial <7,35). La acidosis metabólica puede deberse a un aumento de la producción de ácidos o a la administración exógena de ácidos.
Hiato aniónico: El cálculo del hiato aniónico suele ser útil en el diagnóstico diferencial en la acidosis metabólica (v. tabla 12-9). El hiato aniónico se calcula restando la suma de las concentraciones de Cl y HCO3- de la concentración plasmática de Na+. Las proteínas plasmáticas cargadas negativamente explican la mayor parte del hiato aniónico; las cargas de otros cationes (K, Ca y Mg) y aniones (PO4, sulfato y aniones orgánicos) plasmáticos tienden a equilibrarse. El intervalo aceptado para el hiato aniónico normal es de 12 ± 4 mEq/l. Por otra parte, este intervalo se basa en los rangos normales de las concentraciones de electrólitos medidas con los métodos utilizados en los años setenta. Actualmente la mayoría de los laboratorios clínicos emplean técnicas diferentes; en consecuencia, el intervalo normal del hiato aniónico ha disminuido y puede ser tan bajo como de 5 a 11 mEq/l. Al valorar el hiato aniónico, los médicos deben tener en cuenta su particular intervalo de referencia de laboratorio.

Cuando se añade un ácido al LEC, el HCO3- lo tampona rápidamente según la reacción siguiente:
HA + HCO3- (r) H2CO3 + A- (r) CO2 + 2O + A-
Cuando la acidosis metabólica se debe a la acumulación de ácidos no medidos, como el sulfato en la insuficiencia renal, los cuerpos cetónicos en la cetoacidosis diabética o alcohólica, o el lactato o los agentes tóxicos exógenos como el etilenglicol o los salicilatos, el hiato aniónico está elevado. Si el anión ácido es el cloruro, resultará una acidosis metabólica hiperclorémica. Dado que el cloruro forma parte de la fórmula para calcular el hiato aniónico, la acidosis metabólica hiperclorémica no producirá aumento del hiato aniónico. Las pérdidas renales o extrarrenales de HCO3- producen acidosis metabólica hiperclorémica (hiato no aniónico), puesto que los mecanismos renales retienen cloruro en un intento para conservar el volumen del LEC.
Cuando se produce un aumento del hiato aniónico, se puede inferir la presencia de una o más sustancias que comúnmente conducen a la acidosis. En la tabla 12-9 se enumeran las causas de acidosis metabólica con aumento del hiato aniónico que se encuentran con mayor frecuencia. En la acidosis diabética, la ausencia de insulina (y el exceso de glucagón) conduce a la producción metabólica de cetoácidos -los ácidos acetoacético y b-hidroxibutírico- y de ácido acético por el hígado. Estos cetoácidos explican la acidosis, así como los aniones no medidos. También suele producirse cetoacidosis en la ingestión crónica de alcohol junto con una dieta escasa, debido a la ingesta pobre en hidratos de carbono y a la inhibición de la gluconeogénesis por el alcohol. El diagnóstico de cetoacidosis se confirma por la presencia de cetoácidos en el plasma. Los cetoácidos se detectan generalmente mediante la reacción con el nitroprusiato. El nitroprusiato reacciona con los ácidos acetoacético y acético, pero no con el ácido b-hidroxibutírico. En el alcoholismo, la cetoacidosis es causada predominantemente por el ácido b-hidroxibutírico. A veces los Pacientes con cetoacidosis diabética tienen también una mayor proporción de ácido b-hidroxibutírico debido a un aumento del cociente entre el nicotinamida-adenina dinucleótido reducido y el oxidado (NADH/NAD). Como los métodos habituales de medir los cuerpos cetónicos no miden el ácido b-hidroxibutírico, la prueba del nitroprusiato estándar puede subestimar el grado de cetosis en esos Pacientes.
Otra causa frecuentemente observada de acidosis metabólica con aumento del hiato aniónico es el ácido láctico, que se produce en el metabolismo anaerobio del ácido pirúvico. A través de las vías glucolíticas fisiológicas se producen normalmente niveles bajos de ácido láctico a partir de la glucosa; sin embargo, cuando existe un aumento de la producción de lactato o una disminución de su consumo, el lactato puede acumularse. La hipoperfusión de los tejidos que tiene lugar en el shock conduce a un aumento de la producción de lactato y también a una disminución de su consumo, y es la causa más frecuente de acidosis láctica. La disfunción hepática debida a una mala perfusión hepática o a lesión hepatocelular también pueden producir acidosis láctica debido a la disminución de la conversión del lactato nuevamente en glucosa. El alcoholismo puede causar asimismo acumulación de lactato por un mecanismo similar. La acidosis láctica se produce igualmente en ciertos tumores malignos, en la diabetes mellitus y en el SIDA, y asimismo de manera idiopática.
La insuficiencia renal es también una causa de aumento del hiato aniónico en la acidosis metabólica. Al disminuir la función renal se acumulan varias sustancias en el plasma, como PO4, sulfatos, urato e hipurato. Dado que en otras formas de acidosis metabólica con aumento del hiato aniónico existen a veces grados variables de uremia, un aumento del hiato aniónico sólo debe atribuirse a insuficiencia renal tras una búsqueda minuciosa de otras causas.
La sobredosis de diversas sustancias también causa acidosis metabólica con aumento del hiato aniónico. En la intoxicación por salicilatos, metanol o etilenglicol, la interferencia con el metabolismo intermediario normal y la acumulación de aniones orgánicos exógenos causa acidosis metabólica. La identificación inmediata es crucial para minimizar el daño de los órganos en esos Pacientes. Cuando existe acidosis con un hiato aniónico normal debe sospecharse un deterioro de la excreción renal de ion H+. El deterioro de la excreción renal de ácidos puede deberse a una nefropatía intrínseca, como la acidosis tubular renal (ATR), o a una nefropatía intersticial, o a una respuesta a pérdidas de volumen extrarrenales y de HCO3-. En la ATR se produce alguno de los diferentes defectos tubulares renales específicos de la reabsorción de HCO3- o de la secreción de ion H+, o ambas cosas. La TFG no suele estar afectada. La ATR proximal (tipo 2, ATR con pérdida de bicarbonato) es consecuencia de un defecto en la reabsorción de HCO3- en el túbulo proximal. La ATR proximal se presenta en asociación con glucosuria, fosfaturia y aminoaciduria en niños con el síndrome de Fanconi. Este síndrome se presenta también raras veces en adultos con mieloma múltiple y con el uso de tetraciclina caducada de fecha. Se necesitan grandes dosis de HCO3- para corregir la acidemia en estos Pacientes, porque debido a que el túbulo distal funciona normalmente, los Pacientes con ATR proximal pueden acidificar su orina. La ATR distal (tipo 1, clásica) es consecuencia de un defecto en el mecanismo de acidificación de la nefrona distal. Existen formas primarias, pero son más frecuentes las formas adquiridas. La ATR distal puede presentarse secundariamente a anemia de células falciformes, hipercalcemia, toxicidad por anfotericina B, toxicidad por tolueno (inhalación de pegamentos o pinturas) o toxicidad por litio. Una ATR mucho más común en adultos es el hipoaldosteronismo hiporreninémico (ATR tipo 4). La ATR tipo 4 acompaña a menudo a la diabetes mellitus y a la nefropatía intersticial. La ATR también se presenta debido al daño tubulointersticial causado por la nefropatía de los analgésicos, la pielonefritis crónica y la uropatía obstructiva. El deterioro de la secreción renal de ácido también puede presentarse en la insuficiencia renal aguda o en la crónica avanzada, por lo que una acidosis con hiato aniónico normal también se produce a veces por una insuficiencia renal aislada.
Las pérdidas extrarrenales de HCO3- y de volumen tienen lugar principalmente a través del tracto GI. Las pérdidas excesivas de líquido GI debidas a diarrea prolongada, adenoma velloso del colon o drenaje de líquido biliar, pancreático o intestinal, pueden producir acidosis metabólica, particularmente en presencia de insuficiencia renal. En una derivación del tracto urinario, como en la ureterosigmoidostomía, el Cl de la orina es intercambiado por HCO3- en el colon y también es absorbido el amoniaco urinario. Debido a los problemas con las ITU crónicas y los tumores del asa sigmoidea, la ureterosigmoidostomía se realiza raramente. Los Pacientes con una ureteroileostomía (conductos ileales) o en quienes se ha construido una vejiga ortotópica tienen muchos menos problemas de acidosis metabólica, en especial si la función renal no está deteriorada. Sin embargo, puede presentarse acidosis metabólica si la disfunción del asa o la vejiga producen retención urinaria.
Síntomas, signos y diagnóstico
Los principales síntomas y signos de acidosis suelen estar enmascarados por los de la enfermedad subyacente y son difíciles de separar de ellos. La acidosis leve puede ser asintomática o ir acompañada de una lasitud vaga, náuseas y vómitos. El hallazgo más característico en la acidosis metabólica grave (pH <7,20, HCO3- <10 mEq/l) es el aumento de la ventilación, que se produce como parte de la compensación respiratoria. Al principio existen aumentos poco apreciables de la profundidad de la ventilación. Después puede observarse una mayor frecuencia respiratoria y ventilación con los labios fruncidos (respiración de Kussmaul). También puede haber signos de depleción de volumen del LEC, especialmente en Pacientes con cetoacidosis diabética o pérdidas de volumen GI. La acidosis grave puede causar shock circulatorio debido al deterioro de la contractilidad del miocardio y la respuesta vascular periférica a las catecolaminas; también se produce con frecuencia una obnubilación progresiva.
Datos de laboratorio
En la acidosis metabólica el pH arterial es <7,35 y el HCO3- es <21 mEq/l. En ausencia de enfermedad respiratoria la PCO2 es <40 mm Hg debido a la compensación respiratoria. En la acidosis metabólica simple se puede esperar que la PCO2 disminuya aproximadamente 11 a 13 mm Hg por cada 10 mEq/l de reducción en el HCO3- plasmático. Un descenso de la PCO2 mayor o menor que el esperado sugiere la coexistencia de alcalosis respiratoria primaria o de acidosis respiratoria u otra alteración metabólica primaria, respectivamente.
Cuando la función renal es normal y no existe depleción del volumen, el pH urinario puede caer por debajo de 5,5, con acidosis grave. Un pH urinario con una reducción inferior a la máxima implica disfunción renal debida a insuficiencia renal aguda o crónica, enfermedad tubulointersticial o acidosis tubular renal.
En la cetoacidosis diabética existe casi siempre hiperglucemia; en la mayoría de los casos se puede confirmar la cetonemia mediante la prueba del nitroprusiato. Es preciso tener cuidado al interpretar los resultados de la prueba del nitroprusiato en los casos en los que los niveles de ácido b-hidroxibutírico pueden estar elevados, porque el nitroprusiato no detecta el ácido ß-hidroxibutírico.
La intoxicación por etilenglicol debe sospecharse en los Pacientes con una acidosis inexplicada y cristales de oxalato en la orina. Hiatos aniónicos particularmente altos (20 a 40 mEq/l) se presentan típicamente en la intoxicación por etilenglicol y metanol. Los niveles de ambas sustancias se pueden determinar en el plasma y a veces son útiles cuando el diagnóstico no es obvio a partir de la historia clínica y otros hallazgos. La intoxicación por salicilatos se caracteriza por alcalosis respiratoria poco después de la ingestión y por acidosis metabólica más tardía en la evolución. Se puede recurrir a la determinación de los niveles de salicilato para ayudar en el diagnóstico. La toxicidad está señalada por concentraciones plasmáticas >30 m g/dl (>2,17 m mol/l).
Dado que la depleción de volumen acompaña a la acidosis, es frecuente una azoemia leve (BUN 30 a 60 m g/dl [10,7 a 21,4 mmol urea/l]). Las elevaciones mayores del BUN, especialmente asociadas a hipocalcemia e hiperfosfatemia, sugieren la insuficiencia renal como causa de la acidosis. La hipocalcemia puede presentarse también en asociación con shock séptico. Las alteraciones en el K plasmático durante la acidosis se comentan en otro apartado (v. más atrás Metabolismo del potasio). La hiperpotasemia es relativamente infrecuente en la acidosis láctica a no ser que vaya acompañada de insuficiencia renal y/o un aumento del catabolismo tisular.
Tratamiento
El tratamiento directo de la acidemia con HCO3- resulta tal vez intuitivo, pero el bicarbonato sódico no está claramente indicado en ciertas circunstancias. Cuando la acidosis metabólica es producida por ácidos inorgánicos (es decir, acidosis con hiato aniónico hiperclorémico o normal), el HCO3 es necesario para tratar la alteración acidobásica. Sin embargo, cuando la acidosis se produce por acumulación de ácidos orgánicos (es decir, acidosis con aumento del hiato aniónico), como en la acidosis láctica, la cetoacidosis o los síndromes de intoxicación señalados en la tabla 12-9, el papel del Na+HCO3 es discutible. Los médicos contrarios al tratamiento con Na+HCO3 insisten en que la mortalidad de cada una de esas patologías está relacionada más estrechamente con la gravedad de los procesos de la enfermedad subyacente que con el grado de acidemia. Otros argumentos contra el uso del tratamiento alcalino son la posibilidad de sobrecarga de Na+ y de volumen, la hipopotasemia, la acidosis del SNC, la hipercapnia y la alcalosis por exceso. A la inversa, es conocido que la acidosis se asocia a diversos efectos cardiovasculares perjudiciales, como la reactividad disminuida a los agentes presores. Los Pacientes acidóticos son especialmente vulnerables a nuevos descensos del pH por cambios bastante insignificantes de la concentración plasmática de HCO3-. Los autores que proponen el tratamiento con Na+HCO3 señalan que la acidosis con aumento del hiato aniónico también se presenta con frecuencia asociada a insuficiencia renal y acidosis tubular renal, trastornos en los que el tratamiento con Na+HCO3 no se discute. Independientemente de si se administra Na+HCO3, es preciso identificar la causa subyacente a la acidosis y tratarla siempre que sea posible.
A pesar de estas y otras controversias, la mayoría de los especialistas siguen recomendando un uso prudente del bicarbonato sódico i.v. en el tratamiento de la acidosis metabólica grave (pH <7,20). Esta clase de tratamiento se puede administrar añadiendo cantidades variables de bicarbonato sódico (44 a 88 mEq) a solución de dextrosa al 5% o bien a solución salina hipotónica (0,45%), según la situación clínica y las alteraciones asociadas del agua y el volumen. El objetivo del tratamiento con HCO3- es elevar el pH sanguíneo a 7,20 y el HCO3- plasmático 8 o 10 mEq/l. La cantidad de Na+HCO3 necesaria puede calcularse mediante la fórmula
Na+HCO3 necesario [mEq] = [(HCO3-) deseado - (HCO3-) observado] x 0,4 x peso corporal [kg]
Cuando existe insuficiencia renal, y cantidades incluso moderadas de Na+HCO3 amenazan producir sobrecarga de volumen, puede estar indicada la hemofiltración (combinada con bicarbonato i.v.) o la hemodiálisis contra un líquido dializador añadido con HCO3-. El dicloroacetato potencia la oxidación del lactato y ha sido propuesto como alternativa al Na+HCO3 en el tratamiento de la acidosis láctica. Sin embargo, ensayos controlados han mostrado escasos beneficios con su uso.
Un control más específico de la acidosis metabólica depende de la causa subyacente. El tratamiento de la acidosis láctica es sobre todo de mantenimiento. Si es posible, debe buscarse y contrarrestarse la causa del aumento en la generación de lactato (o de la disminución del aclaramiento del lactato). El control de la cetoacidosis diabética se describe en otro apartado (v. Diabetes mellitus, cap. 13).
La intoxicación con metanol o etilenglicol es una urgencia médica por la toxicidad de los metabolitos de esos compuestos. El tratamiento específico incluye etanol i.v. para inhibir su metabolismo y dar tiempo para el aclaramiento por los riñones. La hemodiálisis se necesita si existe disfunción renal o en caso de intoxicación grave. El tratamiento de la acidosis tubular renal y de la acidosis debida a nefropatía crónica requiere el uso de Na+HCO3. El tratamiento de estos trastornos se comenta más ampliamente en el capítulo 229.
Alcalosis metabólica
Situación con pH arterial elevado, aumento de la concentración plasmática de HCO3- y generalmente hipoventilación alveolar compensadora que produce un aumento de la PCO2.
Etiología y patogenia
La alcalosis metabólica se produce por una pérdida de ácido o una ganancia de álcalis netas en el LEC. Si la alcalosis sobrepasa la capacidad tamponadora del pH sanguíneo se produce alcalemia (pH arterial >7,45). La pérdida de secreciones gástricas de contenido ácido a través de vómitos prolongados o aspiración nasogástrica, las pérdidas excesivas de ácido por la orina o las heces y el movimiento transcelular de iones H+ hacia el interior de las células conducen a una pérdida neta de ácidos del LEC. Una ganancia neta de álcalis puede producirse con la administración excesiva aguda o crónica de álcalis, especialmente en Pacientes con insuficiencia renal o una grave depleción de volumen. Cualquiera que sea la causa, los riñones tienden a corregir con rapidez la alcalosis mediante la excreción rápida del exceso de HCO3-. Para que aparezca una alcalosis metabólica tienen que existir factores que inhiban la excreción renal de HCO3-.
La infusión de Na+HCO3 puede causar alcalosis metabólica sólo si la infusión es rápida y se administra una gran carga de HCO3-. La alcalosis se produce porque la administración de álcalis supera a la excreción de HCO3-; suele ocurrir tras la administración de Na+HCO3 en la RCP. La administración crónica de un exceso de álcalis puede causar también alcalosis metabólica. El síndrome de la leche y los alcalinos se produce por la ingestión crónica de cantidades excesivas de carbonato cálcico. Los hallazgos son hipercalcemia, nefrocalcinosis, insuficiencia renal y alcalosis metabólica. Se cree que la alcalosis metabólica en ese síndrome se debe a la dificultad de la excreción de HCO3- relacionada con la insuficiencia renal (v. más atrás Hipocalcemia).
Si no existe una ingestión excesiva de álcalis, la alcalosis metabólica implica un defecto en la excreción renal de HCO3-. La depleción de volumen, la deficiencia de K y/o el exceso de mineralocorticoides son las situaciones clínicas más frecuentes que inhiben la excreción renal de HCO3-. En la tabla 12-9 se enumeran los trastornos que conducen comúnmente a la alcalosis metabólica.
Tal vez el más frecuente e importante de esos trastornos es la contracción de volumen del LEC. La depleción de volumen (o la depleción de cloruro) conduce a liberación de renina y aldosterona, lo cual causa una ávida reabsorción renal de Na+ en la nefrona distal. Esto lleva a una pérdida subsiguiente de ion H+ y de K, que pueden mantener o empeorar la alcalosis. La pérdida de HCl gástrico por un drenaje nasogástrico o por vómitos prolongados produce al principio una alcalosis metabólica debida a la pérdida neta de ácido. La alcalosis se mantiene por la pérdida concomitante de Cl (y de volumen) y por el desarrollo de una hipopotasemia. El uso excesivo de laxantes también puede causar alcalosis metabólica a través de la pérdida de ácido y de volumen procedente del tracto GI. El abuso subrepticio de laxantes debe sospecharse en Pacientes, por lo demás normales, con hipopotasemia y alcalosis metabólica persistente, especialmente en los que están preocupados por perder peso.
Cuando la hipoventilación (hipercapnia) dura más de unos pocos días, los mecanismos renales compensan la acidosis respiratoria mediante la retención de HCO3-. Cuando la hipercapnia se corrige con rapidez, muy frecuentemente con la ventilación mecánica, puede producirse alcalosis metabólica (alcalosis metabólica poshipercápnica). Este trastorno responde generalmente a la corrección de la ventilación por minuto a un nivel más apropiado para el Paciente. A pesar del retorno a la ventilación adecuada la alcalosis suele persistir hasta que se reponga el Cl (volumen).
Los diuréticos son una causa frecuente tanto de estados sensibles como resistentes al Cl (v. tabla 12-9). Los diuréticos pueden causar una contracción aguda de volumen del LEC e hipermineralocorticoidismo, los cuales son sensibles a la administración de Cl (volumen). Sin embargo, puede presentarse un estado resistente al Cl en un tratamiento diurético continuo debido a las altas pérdidas urinarias de Cl y a la depleción concomitante de K.
La alcalosis metabólica resistente al Cl se puede definir funcionalmente como una alcalosis metabólica que no responde a la administración de Cl (volumen). La deficiencia grave de Mg o K puede causar aumento de la excreción renal de ion H+ y una alcalosis subsiguiente que no responde a la reposición del volumen hasta que se corrigen las deficiencias electrolíticas. En los estados de exceso persistente de esteroides suprarrenales, se produce alcalosis por la reabsorción de Na+ en el túbulo distal mediada por los mineralocorticoides. La reabsorción de Na+ en el túbulo distal conduce a una expansión del volumen del LEC y a una secreción persistente de iones H+ y K. Las causas del aumento de actividad mineralocorticoide son, entre otras, el hiperaldosteronismo primario, el síndrome o la enfermedad de Cushing, los tumores secretores de renina, la estenosis de la arteria renal y los defectos enzimáticos congénitos suprarrenales.
El ácido glicirretínico inhibe la conversión enzimática del cortisol en metabolitos menos activos. Como el cortisol tiene actividad mineralocorticoide intrínseca, la ingestión de sustancias que contienen ácido glicirretínico, como el regaliz y algunos tipos de tabaco de mascar, puede imitar un hipermineralocorticoidismo. Finalmente, los síndromes de Bartter y Gitelman son raros trastornos hereditarios asociados con niveles elevados de renina y de aldosterona y alcalosis metabólica hipopotasémica. El síndrome de Gitelman puede distinguirse del síndrome de Bartter por la presencia de hipomagnesemia e hipocalciuria. El síndrome de Gitelman se presenta sobre todo en adultos jóvenes, mientras que el síndrome de Bartter es más común en la niñez temprana, con pérdidas renales de sal y depleción de volumen.
Síntomas, signos y diagnóstico
Las manifestaciones clínicas más frecuentes de la alcalosis metabólica son la irritabilidad y la hiperexcitabilidad neuromuscular, quizá debidas a hipoxia por un desplazamiento transitorio de la curva de disociación de la oxihemoglobina hacia la izquierda. La alcalemia conduce a un aumento de unión del calcio ionizado a las proteínas, a pesar de que el Ca plasmático esté inalterado; la alcalemia grave puede hacer que el Ca ionizado disminuya lo suficiente como para provocar tetania (v. más atrás Hipocalcemia). Dado que la hipocalcemia suele acompañar a la alcalosis metabólica, también puede haber signos de depleción de K concomitante, como debilidad muscular, espasmos, íleo y poliuria.
Debe sospecharse la presencia de alcalosis metabólica cuando la historia o la exploración física indican depleción de volumen, pérdida de volumen GI crónica o alguna de las situaciones clínicas enumeradas en la tabla 12-9.
Datos de laboratorio
El diagnóstico clínico de la alcalosis metabólica requiere la determinación del HCO3- plasmático y el pH arterial. El pH arterial es >7,45 y el HCO3- >40 mEq/l en la alcalosis metabólica primaria (elevaciones menos llamativas del HCO3- plasmático pueden deberse a la compensación de una acidosis respiratoria crónica). Debido a la hipoventilación compensadora pueden producirse aumentos de la PCO2 tan altos como 50 a 60 mm Hg, especialmente en Pacientes con una insuficiencia renal leve. En la alcalosis metabólica simple se puede esperar que la PCO2 aumente aproximadamente 6 a 7 mm Hg por cada 10 mEq/l de incremento en el HCO3- plasmático. Un aumento de la PCO2 superior o inferior al esperado sugiere la coexistencia de acidosis respiratoria primaria, o de alcalosis respiratoria u otra alteración metabólica primaria, respectivamente.
Además del aumento de HCO3- plasmático, el patrón electrolítico típicamente presente en la alcalosis metabólica incluye hipocloruremia, hipopotasemia y, a veces, hipomagnesemia. Cuando la alcalosis metabólica se asocia con depleción de volumen del LEC, el Cl urinario está casi siempre bajo (<10 mEq/l), mientras que el Na+ urinario puede superar los 20 mEq/l en las fases iniciales. A la inversa, la alcalosis asociada con un exceso primario de esteroides suprarrenales y expansión de volumen se caracteriza por un alto nivel de cloruro en la orina. El pH urinario en la alcalosis metabólica es alcalino excepto en presencia de una grave depleción de K, en este caso puede producirse aciduria paradójica.
Tratamiento
Cuando es leve, la alcalosis metabólica no suele requerir ningún tratamiento específico. El tratamiento más eficaz es la correccion del defecto subyacente que causa el deterioro de la excreción renal de HCO3-. La alcalosis metabólica suele remitir cuando los déficits de cloruro (y de volumen) del LEC son repuestos con solución salina oral o i.v. En el estado poshipercápnico, la alcalosis metabólica persistente responde también a la administración de Cl, con frecuencia en forma de soluciones de Na+Cl o KCl. El ClNa+ debe administrarse con precaución, o evitarse, en los Pacientes propensos a una sobrecarga de volumen. En estos Pacientes el KCl suele ser una alternativa segura, a no ser que exista un deterioro renal grave. La solución diluida de HCl también es eficaz, pero puede producir una hemólisis brusca. También es eficaz el cloruro amónico oral, pero debe evitarse en Pacientes con hepatopatía.
En la deficiencia grave de K, o en Pacientes con hipermineralocorticoidismo, la alcalosis es resistente al Cl y no puede corregirse hasta que haya sido repuesto el K. Es preciso instaurar un tratamiento más específico de la causa subyacente. Los síndromes de Bartter y de Gitelman son frecuentemente difíciles de tratar; la corrección de la hipopotasemia es el pilar principal del tratamiento. La inhibición del eje renina-angiotensina-aldosterona con inhibidores de la ECA ha obtenido un éxito limitado.
Acidosis respiratoria
Situación con pH arterial bajo, hipoventilación que produce una PCO2 elevada y generalmente un aumento compensador de la concentración plasmática de HCO3-.
Etiología y patogenia
La acidosis respiratoria se produce en la depresión del centro respiratorio (debida a fármacos, anestesia, enfermedad neurológica, sensibilidad anormal al CO2), en anomalías del fuelle torácico (poliomielitis, miastenia grave, síndrome de Guillain-Barré, lesiones por aplastamiento del tórax), reducción intensa del área de la superficie alveolar para el intercambio de gases (trastornos caracterizados por desequilibrio ventilación/perfusión como la EPOC, neumonía grave, edema pulmonar, asma o neumotórax) y obstrucción laríngea o traqueal.
El contenido sanguíneo de CO2 es una función de las tasas de producción y de eliminación de CO2. La producción varía con el porcentaje de calorías que se obtienen a partir de los hidratos de carbono, mientras que la tasa de ventilación alveolar controla la eliminación de CO2 por el pulmón. La presión parcial del CO2 gaseoso (PCO2) es directamente proporcional a la cantidad total de CO2 contenida en una solución de sangre. Cualquier aumento en la PCO2 debido a un aumento de producción de CO2 es controlado rápidamente por un aumento de la ventilación alveolar. Análogamente, una disminución de la ventilación alveolar produce retención de CO2 en los pulmones y origina una acidosis respiratoria.
La acidosis respiratoria se asocia con frecuencia con alteraciones neurológicas. Esta clase de alteraciones puede exacerbarse con el desarrollo de acidosis en el LCR o de acidosis intracelular cerebral, hipoxemia concurrente y alcalosis metabólica.
Síntomas, signos y diagnóstico
La alteración más característica es la encefalopatía metabólica con cefalea y somnolencia que evolucionan a estupor y coma. Por lo general se desarrolla lentamente a medida que avanza la insuficiencia respiratoria, pero la encefalopatía abrupta, plenamente desarrollada, puede ser desencadenada por sedantes, una infección pulmonar o una alta fracción de O2 en el aire inspirado (FIO2) en Pacientes con una insuficiencia respiratoria avanzada. También pueden presentarse asterixis y mioclono multifocal. A veces se produce dilatación de las vénulas retinianas y papiledema como consecuencia del aumento de la presión intracraneal. La encefalopatía puede ser reversible si no se presenta daño cerebral por la hipoxia.
Datos de laboratorio
En la acidosis respiratoria aguda, el pH bajo se debe a una elevación aguda de la PCO2. El HCO3- puede estar normal o ligeramente aumentado. Un aumento brusco de la PCO2 se asocia con un incremento del HCO3- plasmático no superior a 3 o 4 mEq/l debido al sistema de tamponamiento celular. Cuando la compensación renal se ha desarrollado completamente, como en la acidosis respiratoria crónica, el descenso del pH es amortiguado, debido a la retención renal de HCO3- y al aumento del HCO3- plasmático. La elevación compensadora esperada en el HCO3- plasmático es aproximadamente de 3 a 4 mEq/l por cada 10 mm Hg de incremento en la PCO2 (v. tabla 12-8). Un aumento del HCO3- plasmático superior o inferior al esperado indica la coexistencia de alcalosis metabólica primaria o de acidosis metabólica primaria, respectivamente.
Tratamiento
Es preciso que el tratamiento corrija la alteración pulmonar subyacente. La insuficiencia respiratoria grave con hipoxemia intensa suele requerir ventilación asistida mecánicamente. Deben evitarse los sedantes (narcóticos, hipnóticos), excepto si son imprescindibles para facilitar la ventilación mecánica. Aunque la mayoría de los Pacientes con retención crónica de CO2 e hipoxia toleran un aumento moderado de la FIO2, algunos responden con un descenso importante del volumen minuto respiratorio y una nueva elevación de la PCO2. Se supone que estos Pacientes se han adaptado a la hipercapnia crónica, por lo cual su principal estímulo respiratorio es la hipoxemia. En consecuencia se debe administrar la concentración de O2 mínima necesaria para elevar la PaO2 a niveles aceptables (>50 mm Hg). Esto se puede realizar administrando el O2 con mascarilla, empezando con una concentración de O2 a un 24%. Debe monitorizarse estrictamente la PCO2; si empieza a aumentar, y en especial si se asocia con alteraciones neurológicas o evidencia de inestabilidad hemodinámica, es preciso considerar la ventilación asistida mecánicamente.
Si se emplea la ventilación mecánica en Pacientes con insuficiencia respiratoria crónica, la PCO2 debe reducirse lentamente, en especial si existe una compensación renal bien establecida o una alcalosis metabólica concomitante, la cual se identifica por un HCO3- alto y un pH normal o alcalino. La reducción rápida de la PCO2 puede causar una alcalosis grave (v. más atrás Alcalosis metabólica). El desplazamiento resultante de la curva de disociación de la oxihemoglobina hacia la izquierda y la vasoconstricción cerebral pueden llevar a convulsiones y a la muerte. Estas consecuencias neurológicas serán evitables proporcionando suficiente O2 inspirado, rebajando la PCO2 con más lentitud y corrigiendo los déficits de K y Cl.
En la acidosis respiratoria que coexiste con alcalosis metabólica primaria o con acidosis metabólica primaria, como ocurre con todas las alteraciones acidobásicas mixtas, el tratamiento correcto reside en la identificación y el tratamiento correctos de cada alteración acidobásica primaria.
Alcalosis respiratoria
Situación con pH arterial elevado, hiperventilación que produce una PCO2 baja y generalmente una disminución compensadora de la concentración plasmática de HCO3-.
Etiología y patogenia
Las causas frecuentes de hiperventilación son la ansiedad (síndrome de hiperventilación), el exceso de ventilación en Pacientes con ventilación asistida, trastornos primarios del SNC, salicilismo, cirrosis hepática, coma hepático, hipoxemia, fiebre, dolor y septicemia por bacterias gramnegativas. La hiperventilación, que lleva a una pérdida excesiva de CO2 en el aire espirado, conduce a alcalosis respiratoria. A medida que descienden la PCO2 plasmática y la PCO2 del tejido cerebral, aumentan el pH plasmático y el cerebral. Se origina vasoconstricción cerebral, y pueden producirse hipoxia cerebral y los síntomas característicos de la hiperventilación.
Síntomas, signos y diagnóstico
La frecuencia y la profundidad de la ventilación suelen aumentar notablemente, en especial cuando la alcalosis respiratoria se debe a trastornos cerebrales o metabólicos. El patrón de ventilación del síndrome inducido por la ansiedad varía desde respiraciones profundas y suspirosas a una ventilación sostenida, manifiestamente rápida y profunda. Los Pacientes suelen quejarse de ansiedad y falta de aliento y dolor torácico. Sorprendentemente estos Pacientes no suelen ser conscientes de su modo de respirar. La principal queja es a veces la incapacidad para recuperar el aliento o tomar suficiente aire, a pesar de estar produciéndose una ventilación excesiva sin dificultades. Pueden presentarse tetania, parestesias periorales, acroparestesias, vértigo o aturdimiento y síncope. En estos Pacientes, los síntomas se pueden reproducir a menudo mediante hiperventilación voluntaria. Los niveles sanguíneos de lactato y piruvato aumentan y el Ca ionizado disminuye. En todas las situaciones, el diagnóstico de hiperventilación se confirma por una PCO2 baja.
Datos de laboratorio
En la alcalosis respiratoria aguda, el rápido descenso de la PCO2 a 20 a 25 mm Hg se asocia con una caída en el HCO3- plasmático no mayor de 3 a 4 mEq/l debido al tamponamiento celular. En la alcalosis respiratoria crónica se puede esperar que el HCO3- plasmático disminuya aproximadamente en 4 a 5 mEq/l por cada 10 mm Hg de reducción de la PCO2 (v. tabla 12-8). Un descenso superior o inferior al esperado en el HCO3- plasmático sugiere la coexistencia de acidosis metabólica primaria o de alcalosis metabólica primaria, respectivamente.
Tratamiento
El principal tratamiento de la alcalosis respiratoria debida a la ansiedad es tranquilizar al Paciente. La reinspiración del CO2 espirado de una bolsa de papel suele ser útil si es posible calmar al Paciente lo suficiente como para ponerle una bolsa en la cara. No deben utilizarse bolsas de plástico porque pueden causar asfixia. Por lo demás, se puede lograr el mismo resultado en cualquier sitio sin necesidad de buscar una bolsa de papel o sentirse cohibido. Se puede decir a la persona que retenga la respiración tanto como pueda, que tome un poco de aire y deje de respirar todo el tiempo posible y repita esta secuencia de 6 a 10 veces. Otras medidas se dirigen a aliviar la ansiedad (v. cap. 187). La hiperventilación con respiradores mecánicos puede corregirse reduciendo la ventilación por minuto o añadiendo espacio muerto. Cuando la hiperventilación se debe a hipoxemia son adecuados la administración de O2 y el tratamiento dirigido a la corrección del intercambio anormal de gas pulmonar. La corrección de la alcalosis respiratoria aumentando la concentración del CO2 inspirado mediante la reinspiración puede ser peligrosa en Pacientes con alteraciones del SNC, dado que esos síntomas pueden estar asociados con un pH bajo del LCR. La PCO2 debe ajustarse con lentitud en estos Pacientes para evitar el desequilibrio entre el pH del LCR y el pH periférico. El tratamiento del salicilismo se comenta en el capítulo 263.