
130 / TRASTORNOS MIELOPROLIFERATIVOS
Grupo de trastornos caracterizados por la proliferación anómala de una o más líneas celulares hematopoyéticas o de elementos del tejido conjuntivo.
Los trastornos mieloproliferativos incluyen la policitemia vera, la mielofibrosis, la leucemia mieloide (mielocítica) crónica (v. cap. 138) y la trombocitemia primaria. Algunos hematólogos también incluyen la leucemia aguda, especialmente la eritroleucemia, y la hemoglobinuria paroxística nocturna; sin embargo, la mayoría argumenta que estos trastornos clonales son suficientemente diferentes de los cuatro básicos y los omiten.
Cada trastorno se identifica según su característica predominante o el lugar de proliferación (v. tabla 130-1). A pesar de la superposición que existe entre ellos, cada uno presenta una constelación típica de características clínicas, datos de laboratorio y evolución. Aunque la proliferación de una línea celular determinada puede dominar el cuadro clínico, los estudios de marcadores citogenéticos e isoenzimas han demostrado que cada trastorno se produce por una proliferación clonal originada en una célula madre pluripotencial, que causa grados variables de proliferación anómala de precursores eritroides, mieloides y megacariocíticos en la médula ósea. Los hematíes, los granulocitos y las plaquetas periféricas se originan a partir de este clon alterado, pero los fibroblastos de la médula ósea no. Los trastornos mieloproliferativos muestran una tendencia variable a terminar en leucemia aguda.
POLICITEMIA VERA
(Policitemia primaria, enfermedad de Vaquez)
Trastorno mieloproliferativo crónico de causa desconocida caracterizado por un aumento de la concentración de Hb y de la masa eritrocitaria (eritrocitosis).
Incidencia y fisiopatología
La policitemia vera (PV) afecta a cerca de 5/1.000.000 personas y predomina en varones (aproximadamente 1,4:1). En el momento del diagnóstico, la edad media es de 60 años (intervalo límite entre 15 y 90 años; raro en la infancia); el 5% de los pacientes tienen menos de 40 años al comienzo de la enfermedad.
En ocasiones, la médula ósea es de aspecto normal, pero generalmente es hipercelular; la hiperplasia afecta a todos los elementos medulares y sustituye a la grasa medular. Existe un aumento de la producción y del recambio de hematíes, neutrófílos y plaquetas. Los megacariocitos también están aumentados y pueden disponerse en acúmulos. El hierro medular está ausente en más del 90% de los pacientes, incluso cuando no se han practicado flebotomías.
El estudio de mujeres con PV heterocigotas para el locus de la G6PD ligado al cromosoma X ha demostrado que los hematíes, los neutrófilos y las plaquetas poseen la misma isoenzima de la G6PD, lo que apoya el origen clonal de esta enfermedad en una célula madre pluripotencial. La causa de esta proliferación se desconoce.
Con el transcurso del tiempo, alrededor del 25% de los pacientes muestran una reducción de la supervivencia eritrocitaria, con imposibilidad de aumentar la eritropoyesis de forma adecuada, desarrollándose anemia y mielofibrosis. En el bazo, el hígado y otras localizaciones aparece hematopoyesis extramedular, con potencial formación de células sanguíneas.
Síntomas y signos
Algunos pacientes permanecen asintomáticos y el diagnóstico se realiza al practicar un análisis rutinario de sangre. Los síntomas (debilidad, cefaleas, mareos, alteraciones visuales, astenia, disnea) generalmente se atribuyen a la expansión del volumen sanguíneo y a la hiperviscosidad. La diátesis hemorrágica es frecuente, al igual que el prurito, particularmente tras baños calientes. La cara puede tener un color rojizo y las venas retinianas estar ingurgitadas. Es habitual la hepatomegalia y más del 75% de los pacientes presentan esplenomegalia (que puede ser masiva, extendiéndose incluso a la cavidad pélvica) y cuando se produce un infarto esplénico, puede auscultarse un roce de fricción. Los pacientes pueden debutar con síntomas de enfermedad ulcerosa péptica, trombosis, síndrome de Budd-Chiari o dolores óseos. Las complicaciones de la hiperuricemia (p.ej., gota, cálculos renales) tienden a aparecer en fases más tardías de la enfermedad.
Finalmente, la actividad eritroide de la médula disminuye. En la sangre periférica se encuentran precursores inmaduros de leucocitos y hematíes y se desarrolla una notable anisocitosis y poiquilocitosis, con microcitos, eliptocitos y hematíes en lágrima. Los neutrófilos y las plaquetas pueden tener un aspecto morfológico alterado y estar elevados sus recuentos. La médula ósea muestra un aumento de las fibras de reticulina y puede aparecer una esplenomegalia progresiva debida a hematopoyesis extramedular. Durante esta «fase gastada» pueden desarrollarse anemia y trombocitopenia.
Las anomalías de la función plaquetaria desencadenan, a menudo, trastornos de la hemostasia. Dado que las intervenciones quirúrgicas pueden constituir un riesgo, la cirugía programada debe posponerse hasta que el Hto sea inferior al 42% y el recuento plaquetario menor de 600.000/ml.
Diagnóstico
La PV debe sospecharse en cualquier varón con un Hto superior al 54% y en cualquier mujer con un Hto mayor del 49%. La PV es una panmielosis, por lo que su diagnóstico es evidente en pacientes con elevación de los tres componentes sanguíneos periféricos, esplenomegalia y sin datos sugestivos de eritrocitosis secundaria. Los criterios diagnósticos se indican en la tabla 130-2.

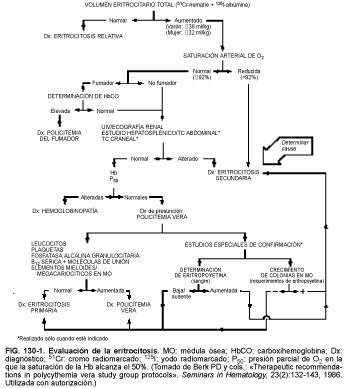
Dado que el Hto es una relación entre el número de hematíes circulantes por unidad de volumen de sangre completa, un Hto elevado puede deberse a una disminución del volumen plasmático. En consecuencia, el diagnóstico de eritrocitosis verdadera se basa en la demostración de un aumento de la masa eritrocitaria. Cuando se determina con hematíes marcados con cromo radiactivo (51Cr), una masa eritrocitaria >36 ml/kg en varones (valor normal, 28,3 ± 2,8 ml/kg) y >32 ml/kg en mujeres (valor normal, 25,4 ± 2,6 ml/kg) se considera anormal. En la eritrocitosis relativa (espúrea) (p. ej., policitemia de estrés, síndrome de Gaisböck), la masa eritrocitaria es normal y el Hto elevado se origina por una disminución del volumen plasmático. Cuando se ha comprobado la existencia de eritrocitosis, debe buscarse su causa (v. tabla 130-3). Es más frecuente la eritrocitosis secundaria (v. más adelante) producida por enfermedades pulmonares, niveles elevados de carboxihemoglobina (policitemia del fumador) y tumores productores de sustancias eritropoyéticas. Las pruebas de laboratorio para el diagnóstico diferencial se muestran en la tabla130-4 y los pasos sugeridos en la evaluación de la eritrocitosis en la figura 130-1.
Si la concentración de O2 de la Hb arterial es inferior al 92%, la hipoxia tisular puede provocar eritrocitosis. El índice de fosfatasa alcalina granulocitaria (FAG) consiste en la tinción histoquímica de una enzima de la población neutrófila. El índice de FAG está elevado en el 75% de los pacientes con PV, pero suele ser normal en los pacientes con otras causas de eritrocitosis. No obstante, debido a que la fiebre, una infección o una inflamación pueden aumentar el índice de FAG, éste sólo tiene valor para confirmar el diagnóstico de PV en ausencia de estos estímulos. El análisis de orina puede revelar hematuria microscópica y la ecografía o la TC renal pueden demostrar una lesión renal responsable de la eritrocitosis secundaria. La P50 (presión parcial de O2 a la que la Hb alcanza una saturación del 50%) mide la afinidad de la Hb por el O2 y se utiliza para excluir una Hb con afinidad elevada (alteración familiar) como causa de la eritrocitosis.


Los pacientes con PV presentan valores de eritropoyetina sérica bajos o indetectables, los que tienen eritrocitosis inducida por hipoxia muestran unos niveles elevados y los individuos con eritrocitosis asociada a tumores tienen valores normales o altos. La médula ósea de los pacientes con PV posee la capacidad de formar colonias eritroides endógenas en cultivos, es decir, es innecesario añadir eritropoyetina. Por el contrario, en personas sanas o en pacientes con eritrocitosis secundaria, la médula requiere la adición de eritropoyetina para formar colonias eritroides.
En la PV pueden aparecer otras anomalías de laboratorio: hiperuricemia e hiperuricosuria en más del 30% de los pacientes, alteraciones cualitativas de la función plaquetaria y elevación frecuente de la vitamina B12 y de su capacidad de fijación.
Pronóstico
Sin tratamiento, el 50% de los pacientes sintomáticos fallecen en los primeros 18 meses tras el diagnóstico. (Para información sobre el apoyo al paciente y su familia, v. cap. 294.) Con tratamiento, la mediana de supervivencia oscila entre 7 y 15 años. La trombosis es la causa más frecuente de muerte, seguida de las complicaciones de la metaplasia mieloide, las hemorragias y el desarrollo de leucemia.
La incidencia de transformación a leucemia aguda es mayor en pacientes tratados con fosfato radiactivo (32P) o agentes alquilantes que en los tratados sólo con flebotomías. La PV que se transforma en leucemia aguda es más resistente a la quimioterapia de inducción que las leucemias de novo.
Tratamiento
La PV es la única forma de eritrocitosis en la que puede estar indicado el tratamiento mielodepresor, por lo que resulta esencial llegar al diagnóstico preciso. El tratamiento debe individualizarse según la edad, el sexo, el estado general, las manifestaciones clínicas y los hallazgos hematológicos.
Las flebotomías son una parte fundamental del tratamiento y pueden ser el único régimen necesario. Es el tratamiento de elección en mujeres en edad fértil y en pacientes menores de 40 años, dado que no son mutagénicas y que hacen desaparecer los síntomas de hipervolemia. Inicialmente, deben eliminarse 300-500 ml de sangre en días alternos hasta alcanzar un Hto menor del 45%. Las flebotomías deben realizarse con más precaución (es decir, 200-300 ml 2/sem) en ancianos y en pacientes con enfermedades cardíacas o cerebrovasculares. Una vez normalizado el Hto, se debe controlar al paciente mensualmente y realizar sangrías si el Hto supera el 45%. La cirugía de urgencia debe ir precedida de la práctica de flebotomías para reducir el volumen eritrocitario a la normalidad. Cuando sea necesario, el volumen intravascular se puede mantener con soluciones cristaloides o coloides.
El tratamiento mielodepresor puede estar indicado en pacientes con recuentos plaquetarios>1 3 106/ml, molestias atribuibles a las visceromegalias, trombosis, síntomas de hipermetabolismo o prurito incontrolado y en ancianos o en pacientes con enfermedad cardiovascular que no toleran bien las flebotomías.
El fosfato radiactivo (32P) proporciona una tasa de éxitos del 80-90%. Las remisiones pueden durar entre seis meses y varios años. Se tolera bien y permite un seguimiento más espaciado cuando se alcanza el control de la enfermedad. No obstante, el 32P se asocia a un incremento de la incidencia de transformación leucémica aguda, por lo que debe realizarse una selección cuidadosa de los pacientes (p. ej., se reserva para pacientes >70años). Tras normalizar el Hto (40-45%) mediante flebotomías, se administra 32P por vía i.v. a dosis de 2,7 mCi/m2 de superficie corporal (dosis total £5 mCi). Esta dosis generalmente normaliza el recuento plaquetario y el Hto en 4-8sem. En caso de no controlarse la enfermedad, puede repetirse la administración de 32P y aumentar la dosis. Si no hay respuesta tras tres inyecciones durante el primer año de terapia, el paciente debe tratarse con flebotomías o con hidroxiurea.
Los agentes alquilantes son leucemogénicos y deben evitarse. Sin embargo, la hidroxiurea, que actúa inhibiendo la enzima ribonucleósido difosfato-reductasa, se ha utilizado con éxito en pacientes en los que está indicado el tratamiento mielodepresor. La hidroxiurea se ha empleado con este propósito durante muchos años; su seguridad a largo plazo con respecto a la posibilidad de leucemogénesis continúa estudiándose. A los pacientes se les practican flebotomías hasta alcanzar un Hto normal (40-45%) y posteriormente reciben hidroxiurea v.o. a dosis de 10-15mg/kg/d. El paciente se controla mediante un hemograma semanal. Cuando se alcanza una situación estable, el intervalo entre hemogramas se alarga a 2 sem y luego a 4 sem. Si el recuento leucocitario se reduce a menos de 4.000/ml o el recuento plaquetario a menos de 100.000/ml, se suspende el tratamiento con hidroxiurea y se reinstaura el 50% de la dosis cuando los recuentos sanguíneos se normalizan. En pacientes con control difícil que requieren sangrías frecuentes o que presentan trombocitosis (recuentos plaquetarios >600.000/ml), puede aumentarse la dosis en 5 mg/ kg/d a intervalos mensuales con controles frecuentes hasta que se estabilice el proceso. La toxicidad aguda es mínima; en ocasiones, los pacientes presentan exantemas, síntomas GI o fiebre.
El interferón-a se ha empleado en pacientes que no toleran la hidroxiurea o en quienes este fármaco no controla el recuento sanguíneo periférico. La dosis de comienzo habitual es de 3,03106 U por vía s.c. 3/sem. El coste, la toxicidad aguda y la seguridad a largo plazo son factores que determinan su uso.
La hiperuricemia puede tratarse con alopurinol v.o. a dosis de 300 mg/d. El prurito se puede tratar con antihistamínicos, pero a menudo es difícil de controlar. Tras el baño, la piel debe secarse suavemente. También se han utilizado con éxito la colestiramina (4 g v.o. 3/d), la ciproheptadina (4-16mg v.o. 4/d) y la cimetidina (300 mg v.o. 4/d). La aspirina alivia los síntomas de la eritromelalgia (dedos de los pies dolorosos e inflamados).
ERITROCITOSIS SECUNDARIA
(Policitemia secundaria)
Tabaquismo. El tabaquismo puede causar eritrocitosis reversible. La carboxihemoglobina es el resultado de la inhalación del humo del tabaco. La eritrocitosis está provocada por anoxia tisular (puesto que la Hb unida al CO no puede transportar O2) y por alteración de la liberación de O2 de la Hb a los tejidos, demostrada por la desviación a la izquierda de la curva de disociación de la oxihemoglobina.
Hipoxemia arterial. Los pacientes con enfermedad pulmonar crónica o cortocircuitos intracardíacos derecha-izquierda con hipoxemia pueden desarrollar eritrocitosis. En la exposición prolongada a grandes alturas (v. cap. 281) o en síndromes de hipoventilación central también puede aumentar la masa eritrocitaria. El tratamiento de los pacientes con enfermedad pulmonar tiene como objetivo mejorar su función pulmonar. Puede requerirse oxigenoterapia y las flebotomías disminuyen la viscosidad y proporcionan una sensación de bienestar al paciente.
Hemoglobinopatías con elevada afinidad por el O2. Este diagnóstico se sospecha cuando existen antecedentes familiares de eritrocitosis y se establece determinando la P50 (v. Diagnóstico en Policitemia vera, arriba) y, si es posible, la curva completa de disociación de la oxihemoglobina. La electroforesis estándar de la Hb generalmente no detecta una banda de Hb anómala y, por tanto, no es fiable para excluir esta causa de eritrocitosis.
Eritrocitosis asociada a tumores. Los tumores y los quistes renales pueden originar eritrocitosis debido al incremento de la secreción de eritropoyetina. La extirpación de la lesión puede ser curativa. Los hepatomas, los hemangioblastomas cerebelosos y los leiomiomas uterinos también pueden causar eritrocitosis paraneoplásica.
MIELOFIBROSIS
(Metaplasia mieloide agnogénica)
Trastorno crónico, generalmente idiopático, que se caracteriza por la fibrosis de la médula ósea, esplenomegalia y anemia leucoeritroblástica, con hematíes en lágrima.
Etiología y patogenia
La causa de la mielofibrosis se desconoce. Puede complicar una leucemia mieloide crónica y se observa en el 15-30% de los pacientes con policitemia vera, si sobreviven el tiempo suficiente. Se han asociado síndromes similares a la mielofibrosis idiopática a diversas neoplasias e infecciones, así como tras la exposición a determinados tóxicos (v. tabla 130-5). La mielofibrosis maligna o aguda, una variante poco frecuente, presenta un curso más rápidamente progresivo y desfavorable; este proceso puede ser, en realidad, una auténtica leucemia megacariocítica.
La incidencia máxima de mielofibrosis idiopática ocurre entre los 50 y los 70 años. La mediana de la supervivencia es de 10 años desde el comienzo aproximado. Los estudios basados en isoenzimas de la G6PD y en anomalías cromosómicas sugieren que se produce la proliferación clonal de una célula madre mieloide anómala. Los fibroblastos medulares no se originan a partir del mismo clon hematopoyético, como se confirma mediante el análisis de los mismos tras el trasplante de médula ósea; por ello se cree que la principal característica de la enfermedad, la mielofibrosis, es un fenómeno reactivo que complica el proceso de la enfermedad primaria.

Síntomas y signos
En las fases precoces, el paciente puede estar asintomático. En un examen rutinario pueden descubrirse esplenomegalia o alteraciones en la sangre. En fases más tardías pueden aparecer malestar general, pérdida de peso y síntomas atribuibles a la esplenomegalia o a infartos esplénicos. En el 50% de los pacientes se halla hepatomegalia. Las adenopatías no son un hallazgo típico, pero pueden encontrarse.
Diagnóstico
Las alteraciones de las células sanguíneas son variables. Es frecuente la anemia que, en general, aumenta con el transcurso del tiempo. Los hematíes son normocrómicos y normocíticos con poiquilocitosis leve, reticulocitosis y policromatofilia. Pueden encontrarse hematíes nucleados en la sangre periférica. En casos avanzados los hematíes presentan formas muy alteradas y en lágrima, siendo su aspecto suficientemente anómalo como para sugerir el diagnóstico.
Los recuentos leucocitarios suelen estar elevados, aunque son muy variables. En la mayoría de los pacientes se encuentran neutrófilos inmaduros y la presencia de mieloblastos no indica necesariamente la conversión a leucemia aguda. La cifra de plaquetas puede estar inicialmente normal, disminuida o aumentada; no obstante, a medida que avanza la enfermedad, existe tendencia a la aparición de trombocitopenia.
El aspirado de médula ósea suele ser seco, requiriéndose la biopsia para demostrar la fibrosis. Como ésta puede no presentar una distribución uniforme, deben realizarse biopsias repetidas en diferentes localizaciones en pacientes sospechosos de padecer mielofibrosis idiopática si la primera biopsia no resulta diagnóstica.
Tratamiento
No existe tratamiento para hacer regresar o controlar el proceso patológico subyacente, aunque se está evaluando la utilidad del interferón-a. El tratamiento se dirige al control de las complicaciones. En algunas ocasiones se han empleado, con fines paliativos, andrógenos, esplenectomía, quimioterapia (hidroxiurea) y radioterapia. En pacientes con concentraciones bajas de eritropoyetina relacionadas con su grado de anemia, la administración de eritropoyetina por vía s.c. puede reducir la necesidad de transfusiones de hematíes. La transfusión de concentrados de hematíes en caso de anemia grave constituye un aspecto fundamental del tratamiento. En pacientes más jóvenes con enfermedad avanzada debe plantearse la realización de un trasplante de médula ósea alogénico.
TROMBOCITEMIA PRIMARIA
(Trombocitemia esencial)
Trastorno caracterizado por aumento del recuento plaquetario, hiperplasia megacariocítica y tendencia hemorrágica o trombótica.
Etiología y patogenia
La trombocitemia primaria es una alteración clonal de una célula madre hematopoyética pluripotencial. Suele aparecer entre los 50 y los 70años y afecta con la misma frecuencia a varones y mujeres. Los recuentos plaquetarios notablemente elevados se deben a un incremento en la producción de plaquetas. La supervivencia de las plaquetas suele ser normal, aunque puede estar disminuida debido a secuestro esplénico. En pacientes ancianos con enfermedad vascular degenerativa, el aumento del número de plaquetas puede desencadenar hemorragias o trombosis graves.
Síntomas y signos
Los síntomas más frecuentes son debilidad, hemorragias, cefalea inespecífica, parestesias de manos y pies y vértigos. Las hemorragias suelen ser leves y se manifiestan por epistaxis, aparición fácil de equimosis o hemorragias GI. Puede observarse isquemia digital y en el 60% de los pacientes hay esplenomegalia que, en general, no se extiende más de 3 cm por debajo del reborde costal izquierdo. También puede existir hepatomegalia.
Diagnóstico
La trombocitemia primaria debe diferenciarse de otros trastornos mieloproliferativos asociados a recuentos plaquetarios elevados. Los requisitos diagnósticos de la trombocitemia primaria incluyen una masa eritrocitaria normal (aumentada en la policitemia vera), ausencia de cromosoma Filadelfia (hallado en la leucemia mieloide crónica) y ausencia de hematíes en lágrima o de un aumento significativo de la fibrosis de la médula ósea (presentes en la mielofibrosis idiopática). El recuento plaquetario puede ser superior a 1 x 1 x 10 106ß/lm, si bien pueden aparecer recuentos bajos de 500.000/ml.
En la extensión de sangre periférica pueden encontrarse agregrados de plaquetas, plaquetas gigantes y fragmentos de megacariocitos. La médula ósea muestra una hiperplasia megacariocítica, con liberación de gran cantidad de plaquetas. En general, los depósitos medulares de hierro son normales.
Tratamiento
Las indicaciones del tratamiento de la trombocitemia primaria son imprecisas, aunque en pacientes con recuentos plaquetarios superiores a 1 x 1 x 106ß/lm y en aquellos con complicaciones hemorrágicas o trombóticas, la mayoría de los autores considera que está indicado el tratamiento definitivo.
El tratamiento mielodepresor consiste en hidroxiurea a dosis de 10-15 mg/kg/d. Es obligatoria la realización semanal de un hemograma. La dosis puede ajustarse como se indicó en el tratamiento de la policitemia vera. También se ha empleado con éxito el fosfato radiactivo (32P) en el tratamiento de la trombocitemia primaria (2,7mCi/m2 i.v.; dosis total £7 mCi). El objetivo del tratamiento es obtener un recuento plaquetario inferior a 600.000/ml sin toxicidad clínica significativa ni disminución del resto de elementos medulares.
En los pacientes con trombocitemia refractaria que requieren tratamiento puede ensayarse la administración de anagrelida, un compuesto imidazoquinazolínico. El tratamiento se inicia con 0,5mg cada 6 h v.o., hasta una dosis total diaria de 2mg. Si no se comprueba disminución (<15% del recuento plaquetario) tras 7 d de tratamiento y la tolerancia al fármaco es adecuada, debe aumentarse la dosis a 1 mg cada 6 h hasta alcanzar una dosis diaria total de 4 mg. Si después de 7-14d el recuento de plaquetas sigue siendo mayor de 600.000/ml y persiste la buena tolerancia, puede incrementarse la dosificación de anagrelida de manera gradual (1-2 mg/d), cada 1-2 sem, hasta que el recuento plaquetario sea inferior a 600.000/ml o bien hasta una dosis diaria total máxima de 12 mg. Es sumamente rara la necesidad de más de 8 mg/d. Deben efectuarse recuentos plaquetarios, al menos, 2/sem en los pacientes tratados por primera vez, ajustando la dosis de anagrelida. Sus efectos secundarios incluyen la disminución de la PA, hipotensión ortostática, insuficiencia renal y molestias gástricas. Se desconocen los riesgos a largo plazo.
Se ha empleado plaquetaféresis cuando se requiere una reducción inmediata del recuento plaquetario (p. ej., casos de hemorragias o trombosis graves o antes de una intervención quirúrgica urgente), debido a que tanto la hidroxiurea como el 32P tardan un tiempo relativamente prolongado (2-6 sem) en tener efecto terapéutico. La aspirina a dosis bajas (es decir, 80 mg/d v.o.) suele utilizarse como fármaco antiplaquetario con el objetivo de prevenir las trombosis, aunque no se ha demostrado que tenga ventajas sobre los resultados obtenidos mediante la reducción aislada del número de plaquetas. También se ha empleado interferón-a, que puede controlar el recuento plaquetario al utilizarlo en terapia continua.

TROMBOCITEMIA SECUNDARIA
La trombocitemia secundaria es un proceso reactivo, cuyas posibles causas se muestran en la tabla 130-6. El recuento de plaquetas suele ser inferior a 1 3 106/ml y la etiología puede resultar evidente a partir de la anamnesis y la exploración clínica; en general, las pruebas de función plaquetaria son normales. No obstante, en el 50% de los pacientes que padecen trastornos mieloproliferativos aparecen alteraciones en la agregación plaquetaria.
El tratamiento de la trombocitemia secundaria es el de la enfermedad subyacente. Con tratamiento, el recuento plaquetario suele normalizarse.