14 /PORFIRIAS
Grupo de trastornos causados por deficiencias de las enzimas de la vía biosintética del hemo.
Se producen niveles anormalmente elevados de porfirinas o sus precursores (p. ej., ácido d-aminolevulínico [ALA] y porfobilinógeno [PBG]), se acumulan en los tejidos y se excretan en la orina y las heces. Las manifestaciones patológicas son producidas casi en su totalidad por los efectos sobre el sistema nervioso y la piel.
Vía de la biosíntesis del hemo
El hemo, un pigmento que contiene hierro, es el componente funcional no proteico de las hemoproteínas, las cuales se encuentran en todos los tejidos.
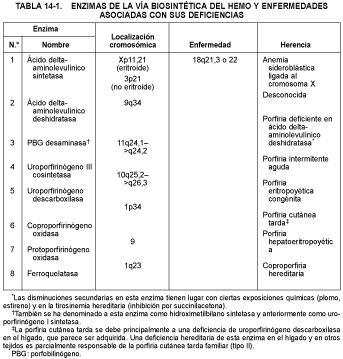
La vía biosintética del hemo se ilustra en la figura 14-1. Las ocho enzimas diferentes que impulsan los pasos secuenciales en esta vía están numeradas de 1 a 8 en la figura 14-1 y se describen brevemente a continuación. La primera enzima y las tres últimas se encuentran en las mitocondrias; las enzimas intermedias están presentes en el citosol.
1. La ALA sintasa, la primera enzima de la vía biosintética del hemo, cataliza la condensación de glicina y succinil-coenzima A para formar ALA. La enzima está localizada en la membrana interna de las mitocondrias y requiere piridoxal-5'-fosfato como cofactor. Genes independientes codifican las ALA-sintasas eritroides y no eritroides.
2. La ALA-deshidratasa, una enzima citosólica, convierte dos moléculas de ALA en un monopirrol, el PBG, con eliminación de dos moléculas de agua. El plomo inhibe la actividad de la ALA-deshidratasa desplazando al cinc (el metal esencial para la actividad enzimática) de la enzima. El inhibidor más potente de la enzima es la succinilacetona, un análogo estructural del ALA, que se encuentra en la orina y la sangre de los Pacientes con tirosinemia hereditaria.
3. La PBG-desaminasa cataliza la condensación de cuatro moléculas de PBG para producir un tetrapirrol lineal, el hidroximetilbilano (HMB). Hay dos isoenzimas de la PBG desaminasa; una está presente exclusivamente en las células eritroides, mientras que la otra se encuentra en células no eritroides. Las dos isoformas de la PBG-desaminasa son codificadas por distintos ARN mensajeros (ARNm) que son transcritos a partir de un solo gen mediante transcripción alternada, corte y acoplamiento.
4. La uroporfirinógeno III cosintetasa cataliza la formación de uroporfirinógeno III a partir del HMB. Esto implica una reordenación intramolecular que invierte la orientación del anillo D (el anillo pirrólico situado en el extremo derecho de la molécula de HMB mostrada en la fig. 14-1) seguida del cierre de un macrociclo para formar uroporfirinógeno III. Cuando esta enzima es deficitaria, el HMB puede experimentar el cierre espontáneo del macrociclo sin la inversión del anillo D, llevando a la formación de uroporfirinógeno I.
5. La uroporfirinógeno descarboxilasa, una enzima citosólica, cataliza 4 descarboxilaciones consecutivas de las cadenas laterales carboximetílicas del uroporfirinógeno III (una porfirina octocarboxílica) para producir heptacarboxilporfirina, hexacarboxilporfirina, pentacarboxilporfirina y, finalmente, coproporfirinógeno III (una porfirina tetracarboxílica). Esta enzima puede metabolizar también el uroporfirinógeno I a coproporfirinógeno I.
6. La coproporfirinógeno oxidasa, una enzima mitocondrial en las células de los mamíferos, cataliza la eliminación del grupo carboxilo y de dos hidrógenos de los grupos propiónicos de los anillos pirrólicos A y B del coproporfirinógeno III para formar grupos vinilos en esas posiciones, formando protoporfirinógeno. Esta enzima no puede metabolizar el coproporfirinógeno I.
7. La protoporfirinógeno oxidasa media en la oxidación de protoporfirinógeno IX a protoporfirina IX, catalizando la eliminación de seis átomos de hidrógeno del núcleo del protoporfirinógeno.
8. La ferroquelatasa cataliza la introducción del hierro en la porfirina, lo que representa el paso final en la vía biosintética del hemo. La enzima no es específica del hierro y puede catalizar la introducción de algunos otros metales, como el cinc.
Los intermediarios de la vía se conservan en el interior de las células y por ello sólo se excretan normalmente en cantidades pequeñas. Ellos difieren notablemente unos de otros en el tamaño de la molécula, la solubilidad y otras propiedades. El ALA, el PBG y los porfirinógenos (las hexahidroporfirinas, es decir, las porfirinas en estado reducido químicamente) son incoloros y no fluorescentes. La protoporfirina, el intermediario final en la vía, es el único producto intermediario que es una porfirina oxidada. Las porfirinas en estado oxidado son de color rojizo y fluorescen cuando son expuestas a una luz ultravioleta de longitud de onda larga. Los porfirinógenos que difunden hacia el líquido extracelular experimentan una autooxidación y son excretados principalmente en forma de porfirinas. No obstante, pueden excretarse en la orina cantidades apreciables de coproporfirinógeno no oxidado. Las moléculas de ALA, PBG, uroporfirina y las porfirinas heptacarboxílicas, hexacarboxílicas y pentacarboxílicas son hidrosolubles y se excretan sobre todo en la orina. La coproporfirina (una porfirina tetracarboxílica) se excreta en la orina y la bilis. La harderoporfirina (una porfirina tricarboxílica) y la protoporfirina (una porfirina dicarboxílica) son escasamente solubles en agua y por ello no pueden ser excretadas por los riñones. Si éstas se acumulan en la médula ósea o el hígado, aparecen en el plasma, son captadas por el hígado y se excretan en la bilis y las heces.
Control de la biosíntesis del hemo
El hemo es sintetizado en cantidades máximas por la médula ósea, donde es incorporado en la hemoglobina, que es una proteína transportadora de oxígeno, y en el hígado, donde la mayoría es incorporado a los citocromos, que son proteínas transportadoras de electrones. Los más abundantes en el hígado son los citocromos P-450, enzimas que metabolizan los fármacos y muchos otros productos químicos extraños al organismo y endógenos (v. cap. 43).
La biosíntesis del hemo está controlada de manera distinta en el hígado y en la médula ósea. La biosíntesis del hemo en el hígado tiene una velocidad limitada y está regulada principalmente por la primera enzima, la ALA-sintasa (v. fig. 14-1, enzima 1). Esta actividad enzimática en las células hepáticas es normalmente muy baja, pero aumenta espectacularmente mediante inducción de la síntesis de la enzima cuando el hígado produce más hemo. La síntesis de la enzima está también bajo un sensible control por retroalimentación por el contenido de hemo libre en las células y disminuye cuando el contenido de hemo libre es alto. Ciertos fármacos y hormonas inducen a los hepatocitos a producir más ALA-sintasa, hemo y citocromo P-450.
En la médula ósea, el hemo se produce en los eritroblastos y los reticulocitos que todavía contienen mitocondrias, mientras que los eritrocitos circulantes carecen de mitocondrias y no pueden formar hemo. La biosíntesis del hemo en las células eritroides está regulada, al menos en parte, por el proceso de captación de hierro en las células. La médula ósea puede expresar las formas específicas de las células eritroides de algunas enzimas de la vía. La ALA-sintasa eritroide-específica está regulada por un elemento sensible al hierro del ARNm, y esto explica en parte la regulación específica de tejido de la síntesis de hemo para la formación de hemoglobina.
Etiología y patogenia
Los genes correspondientes a las ocho enzimas de la vía biosintética del hemo han sido clonados y secuenciados y se han identificado sus localizaciones cromosómicas (v. tabla 14-1). En la forma eritroide-específica de la ALA-sintasa se han encontrado mutaciones en algunos casos de anemia sideroblástica ligada al cromosoma X. Las porfirias y otros trastornos afines están asociados con deficiencias de las otras siete enzimas, y se han caracterizado detalladamente mutaciones de los genes para esas enzimas. Aunque cada tipo de porfiria hereditaria se asocia con la deficiencia de una enzima concreta, es probable que existan diferentes mutaciones en el gen correspondiente a esa enzima, salvo en el caso de que la deficiencia de la enzima proceda de un individuo de la misma familia. Por consiguiente, estas enfermedades son heterogéneas desde el punto de vista molecular.
Cuando una enzima de la síntesis de hemo es deficiente, su substrato y otros precursores del hemo pueden acumularse en la médula ósea o el hígado. Estos precursores aparecen después en exceso en la sangre, son transportados a otros tejidos y se excretan en la orina y las heces.
Algunas porfirias, en especial las que incrementan los precursores iniciales ALA y PBG, lesionan los nervios y conducen a diversos síntomas, como dolor abdominal y debilidad muscular, la cual puede incluso evolucionar a parálisis. Se han postulado mecanismos para las alteraciones neurológicas, tales como los efectos del exceso de intermediarios de la vía del hemo, o la deficiencia de la síntesis de hemo, en el sistema nervioso. No se ha demostrado que el ALA y otros productos de la vía biosintética del hemo sean neurotóxicos, y la deficiencia de hemo en el sistema nervioso tampoco se ha probado. Por tanto, la causa exacta sigue siendo oscura.
Las porfirias que elevan la cantidad de porfirinas como la uroporfirina, la coproporfirina y la protoporfirina en los tejidos y en el plasma originan fotosensibilidad. Cuando se iluminan las porfirinas a longitudes de onda de unos 400 nm en presencia de O2, generan una forma del oxígeno inestable cargada, denominada oxígeno singlete, que puede dañar los tejidos. La piel es particularmente susceptible porque es el tejido más expuesto a la luz.
Clasificación
Las porfirias se clasifican con más precisión en función de las deficiencias enzimáticas específicas. Otras clasificaciones se basan en las manifestaciones clínicas principales; éstas son de utilidad clínica pero pueden solaparse. Las porfirias agudas causan síntomas neurológicos que son generalmente intermitentes. Las porfirias cutáneas causan fotosensibilidad de la piel. La porfiria intermitente aguda, la porfiria por deficiencia de ALA-deshidratasa, la coproporfiria hereditaria y la porfiria variegata constituyen el grupo de las porfirias agudas. La porfiria cutánea tarda, la coproporfiria hereditaria, la porfiria variegata, la protoporfiria eritropoyética, la porfiria eritropoyética congénita y la porfiria hepatoeritropoyética congénita constituyen el grupo de las porfirias cutáneas. En las porfirias hepáticas y eritropoyéticas, el exceso de precursores se origina principalmente en el hígado y en la médula ósea, respectivamente.
En este capítulo se comentan en primer lugar las tres porfirias más frecuentes en el orden que ocupan las enzimas deficientes en la vía de la biosíntesis del hemo (v. también tabla 14-2). Las porfirias menos prevalentes se exponen por separado y con mayor brevedad, también en el orden de las enzimas deficientes en la vía biosintética del hemo.
Pruebas de laboratorio
Los síntomas de las porfirias se parecen a los de muchas otras enfermedades. Algunas pruebas de laboratorio son sensibles y específicas para diagnosticar las porfirias, y los resultados son intensamente anormales cuando esas enfermedades están en actividad. Sin embargo, es preciso elegir e interpretar correctamente las pruebas para obtener una información diagnóstica específica cuando se sospecha la presencia de una porfiria. Lo mejor es apoyarse en unas pocas pruebas sensibles y específicas para la detección selectiva (v. tabla 14-3). En la mayoría de las situaciones, las pruebas selectivas para una porfiria aguda pueden limitarse a la determinación de ALA y PBG en la orina. La prueba de Watson-Schwartz es cualitativa y todavía se utiliza ampliamente para detectar el exceso de PBG urinario. Un método mucho más preferible para la detección rápida del exceso de PBG en orina es utilizar una técnica que emplea una resina de intercambio aniónico colocada en una jeringa de plástico. El método cuantitativo de la columna iónica de Mauzerall y Granick debe utilizarse para verificar los resultados positivos de una prueba selectiva para el PBG y detectar el exceso de ALA. Cuando se sospecha la presencia de una porfiria cutánea se puede determinar la porfirina en el plasma. Las determinaciones de las enzimas eritrocitarias son una segunda línea de pruebas que no se llevan a cabo rutinariamente a no ser que una de las pruebas de detección selectiva sea anormal. Las determinaciones de las porfirinas urinarias, fecales y eritrocitarias se consideran más bien como pruebas de segunda línea; esas pruebas carecen de especificidad (es decir, son también anormales en otros trastornos) y por ello no son muy prácticas para la detección selectiva (v. tabla 14-3).

PORFIRIAS MÁS FRECUENTES
Las tres porfirias más frecuentes son la porfiria intermitente aguda, la porfiria cutánea tarda y la protoporfiria eritropoyética, las cuales se deben a deficiencias de las enzimas tercera, quinta y octava de la vía biosintética del hemo, respectivamente. Estas enfermedades son distintas y difieren considerablemente en sus síntomas, enfoques diagnósticos y tratamientos. La tabla 14-2 compara las principales características de estas tres enfermedades. La porfiria intermitente aguda y la porfiria cutánea tarda comparten características con algunas porfirias menos frecuentes.
PORFIRIA INTERMITENTE AGUDA
(Porfiria aguda intermitente; porfiria sueca; pirroloporfiria)
Trastorno autosómico dominante, es la porfiria aguda más frecuente en la mayoría de los países y se debe a una deficiencia de PBG-desaminasa (HBM-sintasa).
La actividad de la PBG-desaminasa (v. fig. 14-1, enzima 3) es generalmente alrededor de un 50% de la normal en todos los tejidos de los Pacientes con porfiria intermitente aguda (PIA). La mayoría de los Pacientes que heredan este rasgo nunca desarrollan síntomas y se dice que tienen PIA latente.
Incidencia
La PIA se encuentra en todas las razas, pero es algo más frecuente en el norte de Europa. La prevalencia de la PIA y las demás porfirias agudas en Estados Unidos y en la mayoría de los demás países está probablemente alrededor de 5/100.000. La prevalencia puede ser más alta en poblaciones psiquiátricas. El trastorno se manifiesta clínicamente después de la pubertad y más comúnmente en mujeres que en varones; en algunas mujeres, los ataques se producen durante la segunda mitad del ciclo menstrual. Se han descrito unos pocos casos homocigóticos de PIA con síntomas que empiezan en la niñez.
Factores desencadenantes
El trastorno se desencadena por factores externos, como hormonas, fármacos y la dieta. Entre los muchos fármacos implicados están los barbituratos, otros fármacos anticonvulsivos y antibióticos sulfonamídicos (v. tabla 14-4). También pueden desencadenar los síntomas las dietas bajas en calorías y bajas en hidratos de carbono, las cantidades grandes de alcohol y la progesterona y esteroides afines. La mayoría de los fármacos y las hormonas que son perjudiciales en ésta y en otras porfirias agudas inducen la ALA-sintasa hepática y las enzimas de los citocromos P-450. En ocasiones está implicado el estrés debido a infecciones, así como otras afecciones, tratamientos quirúrgicos y problemas psicológicos. Un ataque suele deberse a múltiples factores, algunos de los cuales son a menudo inidentificables.

Síntomas y signos
Los síntomas se presentan en forma de ataques, que evolucionan a lo largo de horas o días y pueden prolongarse durante días, semanas o incluso más tiempo. Los síntomas se deben a los efectos sobre el sistema nervioso; la piel no está afectada.
El dolor abdominal, el síntoma más frecuente, puede ser tan grave que se considere erróneamente como un abdomen quirúrgico agudo. Otros síntomas abdominales son náuseas, vómitos, estreñimiento y diarrea. Puede aparecer distensión del abdomen debida a íleo paralítico. Las manifestaciones abdominales son debidas a los efectos sobre los nervios viscerales. La vejiga puede afectarse de forma análoga y se ha observado retención de orina, incontinencia, disuria y polaquiuria. Dado que no existe inflamación, el dolorimiento a la presión y al soltar la presión sobre el abdomen no son destacados, y la temperatura corporal es normal o ligeramente aumentada. Por consiguiente, los hallazgos en una exploración física pueden ser insignificantes comparados con la intensidad de los síntomas.
Es frecuente la presencia de taquicardia, hipertensión, diaforesis y nerviosismo; estos síntomas pueden deberse a los efectos sobre el sistema nervioso autónomo y a niveles excesivos de catecolaminas en la sangre. La neuropatía motora es frecuente, especialmente en los ataques graves o prolongados, e indica un daño a los axones de los nervios motores. La debilidad muscular suele iniciarse en los hombros y los brazos y puede afectar a todas las neuronas motoras, incluidas las de los pares craneales. Pueden producirse parálisis graves, insuficiencia respiratoria y, rara vez, la muerte. Puede haber temblores y convulsiones. Otras manifestaciones del SNC son síntomas psiquiátricos, como agitación y alucinaciones. La secreción inadecuada de ADH, debida presumiblemente a afectación del hipotálamo, puede originar retención de agua e hiponatremia y puede contribuir a las crisis convulsivas. La recuperación de un ataque puede producirse en unos pocos días, pero la intensa debilidad muscular puede persistir durante meses o años, especialmente si el diagnóstico y el tratamiento se retrasan. La hipertensión puede persistir y estar asociada con deterioro renal. Las anomalías crónicas del hígado son frecuentes y existe un aumento de riesgo inexplicado de carcinoma hepatocelular.
Diagnóstico
Los ataques con síntomas graves abdominales y neurológicos imitan muchas otras enfermedades más frecuentes. Por ello, la PIA y otras porfirias agudas se sospechan y se descartan con más frecuencia que se confirman. Los niveles de ALA y PBG en orina o plasma son muy altos durante los ataques (el PBG urinario oscila generalmente entre 50 y 200 mg/d [221 a 884 mmol/d], intervalo de referencia de 0 a 4 mg/d [0 a 17,7 mmol/d]; y el de ALA 20 a 100 mg/d [145,2 a 726,2 mmol/d], intervalo de referencia de 0 a 7 mg/d [0 a 53,4 mmol/d]) y siguen siendo altos en Pacientes con ataques repetidos. Aumentos de esa magnitud son virtualmente diagnósticos de una de las porfirias agudas, mientras que unos resultados normales en el momento de los síntomas o cerca de él excluye ciertamente las porfirias agudas. El ALA y el PBG son incoloros. Sin embargo, el PBG en solución concentrada forma uroporfirina por un mecanismo no enzimático y además se degrada a sustancias llamadas porfobilinas. El ALA que es hiperproducido en el hígado puede ser metabolizado a porfirinas en otros tejidos. La orina puede ser de color rojizo o pardo debido al exceso de porfirinas o porfobilinas, respectivamente, en especial después de la exposición a la luz. Pero dado que muchas otras sustancias en la orina cambian de color en reposo, un diagnóstico no puede basarse sencillamente en el color de la orina. Las porfirinas fecales suelen estar normales o ligeramente aumentadas, lo cual distingue esta enfermedad de la coproporfiria hereditaria y de la porfiria variegata. La uroporfirina urinaria y la coproporfirina y la protoporfirina eritrocitaria pueden estar aumentadas, pero esos hallazgos no son específicos. En contraste con la porfiria variegata, las porfirinas plasmáticas son normales o sólo están ligeramente aumentadas en la PIA. El intento de aumentar el PBG con fines diagnósticos mediante una carga de glicina o la administración de un fármaco inductor como el fenobarbital puede ser peligroso y no es definitivo.
La PBG-desaminasa eritrocitaria es alrededor de un 50% de la normal en la mayoría de los Pacientes con PIA. Este hallazgo ayuda a confirmar el diagnóstico en Pacientes con aumento del PBG urinario o plasmático. Sin embargo, la determinación de PBG-desaminasa eritrocitaria no es de utilidad como primera prueba en los Pacientes gravemente enfermos con sospecha de tener una PIA, por varias razones: 1) Los intervalos para Pacientes con PIA e individuos normales se superponen en parte. 2) La actividad de la enzima es alta en los eritrocitos jóvenes y disminuye durante los 120 d de plazo vital de los eritrocitos circulantes. Por consiguiente, la enzima puede estar aumentada falsamente cuando concurre la existencia de hemólisis o cuando otro motivo de aumento de la eritropoyesis origina que los eritrocitos sean, en promedio, más jóvenes de lo normal. 3) Algunas mutaciones causantes de PIA están en una región particular del gen de la PBG-desaminasa (dentro o cerca del 1.º de sus 15 exones) de tal modo que la forma de la enzima específica de las células eritroides es normal, mientras que la enzima es deficiente en todos los tejidos no eritroides, incluido el hígado. 4) Las diferencias metodológicas entre laboratorios pueden influir en la especificidad de la prueba, y los problemas en el procesamiento y el transporte pueden conducir a valores falsamente bajos. 5) La enzima no es deficiente en las demás porfirias agudas, la cuales son también importantes a considerar cuando se sospecha una PIA. 6) Un valor bajo de la PBG-desaminasa eritrocitaria en un Paciente con síntomas que sugieren porfiria no confirma que los síntomas se deban a la porfiria, porque la enzima puede estar baja en una PIA clínicamente latente, así como en una PIA activa.
Determinar la PBG-desaminasa eritrocitaria es útil en la detección selectiva de familiares de los Pacientes una vez que en el caso índice de la familia se haya confirmado que tiene un valor bajo de la enzima. A diferencia de los Pacientes con síntomas recientes, los familiares con valores bajos de PBG-desaminasa eritrocitaria, pero que nunca tuvieron síntomas, es improbable que experimenten un aumento del PBG urinario. Durante la detección selectiva de familiares de los Pacientes se debe medir tanto la PBG-desaminasa eritrocitaria como el PBG urinario, porque ninguna de las dos pruebas puede detectar por sí sola de un modo fiable a todos los portadores del rasgo genético.
Los estudios del ADN son los medios más sensibles y específicos para detectar a los familiares que han heredado una mutación asociada con la PIA, pero esos estudios sólo son posibles si se ha identificado previamente la mutación exacta en el caso índice. El diagnóstico in utero es posible, pero está indicado rara vez debido a la favorable perspectiva de la mayoría de los individuos con deficiencia de PBG-desaminasa.
Tratamiento, prevención y pronóstico
El tratamiento de todas las porfirias agudas es en esencia idéntico. Los ataques agudos suelen exigir hospitalización para el tratamiento de los síntomas (v. más adelante). Se observa a los Pacientes en busca de complicaciones neurológicas. Los ataques graves se tratan con hemo, que sólo puede administrarse por vía intravenosa. La pauta estándar es de 3 mg/kg de peso corporal diariamente durante 4 d. El hemo es captado en el hígado, donde suprime la síntesis de la enzima ALA-sintasa limitante de la velocidad y reduce inmediatamente los niveles sanguíneos y urinarios de ALA y PBG. Los síntomas remiten generalmente en varios días. Si se retrasa el tratamiento con hemo, el daño nervioso es más avanzado y la recuperación es más lenta y puede ser incompleta.
En Estados Unidos se dispone de hemo para administración i.v. en forma de hematina liofilizada (hidróxido de hemo) para ser reconstituida con agua estéril. La hematina es inestable cuando se reconstituye de ese modo, y se forman con rapidez productos de degradación, que causan con frecuencia flebitis en el lugar de la infusión y un efecto anticoagulante transitorio. En consecuencia, se recomienda generalmente que la hematina sea estabilizada reconstituyéndola con albúmina humana (para formar albuminato de hemo). El arginato de hemo es un producto del hemo que es estable en forma de solución concentrada y se diluye con solución salina estéril para uso i.v.; se comercializa en algunos otros países, pero no en Estados Unidos. Rara vez el albuminato y el arginato de hemo están asociados con la flebitis, y tampoco causan efectos coagulantes.
El tratamiento con hemo debe iniciarse pronto, pero sólo después de haber confirmado el ataque de porfiria con un intenso aumento del PBG urinario. Después de al menos varios días de tratamiento con hemo, el diagnóstico es más difícil, debido a la reducción inmediata de los niveles de PBG.
Es importante el tratamiento de los síntomas. El dolor se controla con analgésicos narcóticos. Las náuseas, los vómitos, la ansiedad y la intranquilidad se tratan con dosis pequeñas a moderadas de una fenotiazina. (Tras la recuperación, raras veces está indicado el tratamiento continuado con fenotiazina.) Para el insomnio se puede utilizar el hidrato de cloral. Probablemente las benzodiacepinas de acción corta en dosis bajas son seguras para una sedación suave. (Sin embargo, los barbituratos y muchos otros fármacos no lo son.) La distensión vesical puede necesitar un sondaje. Deben suspenderse los fármacos nocivos (v. tabla 14-4), y se deben identificar y corregir si es posible otros factores que contribuyen al ataque.
La ingesta oral se tolera mal, o incluso está contraindicada, a causa de la distensión y el íleo; puede ser necesaria la glucosa i.v. (300 g/d) o una nutrición parenteral más completa. La hospitalización puede no ser imprescindible en Pacientes con ataques recurrentes constantemente leves.
Algunos Pacientes con porfirias agudas desarrollan dolor crónico y otros síntomas continuos. En esos Pacientes, el tratamiento con hemo y la carga de hidratos de carbono no son eficaces.
El tratamiento de las crisis convulsivas crea problemas porque prácticamente todos los fármacos anticonvulsivos (excepto los bromuros y tal vez la gabapentina) pueden exacerbar las porfirias agudas. Las convulsiones durante los ataques pueden ser una consecuencia directa de la porfiria, o pueden ser secundarias a la hiponatremia. En algunos Pacientes coexisten la porfiria y las convulsiones idiopáticas. Si se piensa que las convulsiones están relacionadas con un ataque agudo, los fármacos anticonvulsivos pueden interrumpirse cuando se produzca la recuperación.
Los b-bloqueantes pueden controlar la taquicardia y la hipertensión en los ataques de porfiria aguda, pero pueden ser peligrosos en Pacientes hipovolémicos, en quienes el aumento de la secreción de catecolaminas puede ser un importante mecanismo compensador.
La prevención de los ataques porfíricos es importante, tiene que ser individualizada e incluye lo siguiente: 1) Debe investigarse sistemáticamente a los miembros de la familia para detectar los rasgos latentes y adoptar medidas preventivas. 2) Deben evitarse los fármacos nocivos (v. tabla 14-4). 3) Deben evitarse las dietas estrictas e incluso los períodos de inanición cortos (p. ej., en un postoperatorio o durante enfermedades intercurrentes). Los regímenes dietéticos para la obesidad deben procurar una pérdida de peso gradual durante los períodos de remisión clínica de la porfiria. 4) El tratamiento con hemo puede prevenir muchas veces los ataques recurrentes, pero no existe una pauta estándar. Los estudios actuales indican que puede ser efectiva una sola infusión de hemo una o dos veces por semana. 5) Los frecuentes ataques premenstruales se pueden prevenir con un análogo de la hormona liberadora de gonadotropina y reposición de estrógenos a dosis bajas, pero este uso está todavía en fase de investigación. Los anticonceptivos orales se usan a veces con éxito, pero existe el riesgo de que la progestina llegue a exacerbar la porfiria. Dado que la ovariectomía es irreversible, no debe entrar en consideración para la prevención de los ataques cíclicos salvo que exista otra indicación clínica.
El pronóstico de los Pacientes con deficiencia de PBG-desaminasa es excelente. La gran mayoría nunca desarrolla síntomas, especialmente si tienen concentraciones urinarias normales de precursores de las porfirinas. Aunque estos individuos son menos sensibles a los fármacos inductores que los Pacientes con síntomas porfíricos previos y ALA y PBG persistentemente elevados, deben adoptar las mismas precauciones que los que han tenido ataques porfíricos. La PIA latente no debe considerarse como un riesgo para la salud que limite el acceso de la persona a los seguros sanitarios y de vida.
El pronóstico de los Pacientes que padecen ataques ha mejorado en los últimos 20 años. Los ataques de porfiria son actualmente pocas veces mortales debido al diagnóstico más temprano, a un mejor tratamiento y a la identificación y supresión de los factores incitantes. Aunque en algunos Pacientes se producen ataques de porfiria recurrentes e invalidantes, estos ataques no ocurren durante toda la vida adulta, y la evolución de la enfermedad pocas veces es progresiva.
PORFIRIA CUTÁNEA TARDA
(Porfiria sintomática; porfiria cutánea sintomática; porfiria idiosincrásica)
La más frecuente de todas las porfirias, debida a una deficiencia de uroporfirinógeno descarboxilasa.
La marca específica de la porfiria cutánea tarda (PCT) es la fotosensibilidad, con formación crónica de ampollas en la piel expuesta a la luz solar. La mayoría de los Pacientes con PCT no parecen tener mutaciones en el gen de la uroporfirinógeno descarboxilasa (v. fig. 14-1, enzima 5). La enzima está considerablemente disminuida, pero sólo en el hígado, en estos Pacientes, a los que a veces se asigna una PCT tipo I. Probablemente la deficiencia de la enzima hepática es adquirida, aunque no se ha establecido el mecanismo y todavía se considera posible que tenga una base genética. Una minoría de Pacientes (hasta un 20%) tienen una deficiencia hereditaria de uroporfirinógeno descarboxilasa tal que la enzima está en una proporción aproximadamente la mitad de la normal en todos los tejidos desde el nacimiento. Aunque se dice que estas personas tienen PCT tipo II, no difieren clínicamente de las personas con el tipo I, salvo en que la aparición de los síntomas puede ser más temprana y otros familiares pueden haber estado afectados por el mismo trastorno. En los tipos I y II son importantes los mismos factores desencadenantes y el tratamiento es idéntico. Antes de que los tipos I y II de PCT se manifiesten clínicamente, la actividad de la uroporfirinógeno descarboxilasa tiene que ser mucho menor de la normal en el hígado. Además de la deficiencia de la enzima, la oxidación de sus sustratos (uro-, hepta-, hexa- y pentacarboxilporfirinógeno) a sus correspondientes uroporfirinas puede ser un mecanismo coadyuvante en la PCT.
Epidemiología
Aunque probablemente la PCT es la más frecuente de todas las porfirias, su prevalencia no se ha calculado de manera fiable. La enfermedad se ha identificado en todo el mundo y en todas las razas. Es más frecuente en los varones, pero su frecuencia en las mujeres ha aumentado por el consumo creciente de anticonceptivos orales, estrógenos en la posmenopausia y alcohol. En algunas áreas, proporciones tan grandes como un 80% de los casos se asocian con infección crónica por hepatitis C.
Factores desencadenantes
Los factores que contribuyen a la PCT son en su mayor parte diferentes de los que exacerban la porfiria intermitente aguda. Son entre otros el hierro (en cantidades normales o aumentadas), el alcohol, el virus de la hepatitis C, los estrógenos y a veces los hidrocarburos clorados (p. ej., hexaclorobenceno). Tal vez está asociado el tabaco. Una asociación menos común es la del VIH. Estos factores, en especial unidos al hierro, pueden originar formas del oxígeno lesivas para el hígado que inactivan la uroporfirinógeno descarboxilasa u oxidan su substrato. Se acumulan cantidades masivas de porfirinas en el hígado y son transportadas en el plasma sanguíneo hacia la piel.
Síntomas y signos
Aparecen vesículas y ampollas en las áreas expuestas a la luz solar, como la cara, los brazos y el dorso de las manos. La piel, especialmente la de las manos, es también friable y vulnerable al menor traumatismo. Sigue la formación de costras y cicatrices. La cicatrización es lenta y va seguida a menudo de hiperpigmentación e hipopigmentación, hipertricosis (especialmente facial) y alteraciones seudoesclerodérmicas. Suele producirse lesión hepática y puede deberse en parte a las porfirinas, a infección crónica por el virus de la hepatitis C o al exceso de alcohol. La histopatología del hígado presenta frecuentemente siderosis, transformación adiposa, necrosis y cambios inflamatorios crónicos. Con el tiempo aparecen cirrosis y carcinoma hepatocelular.
En la PCT los depósitos de hierro son normales o están aumentados. Las personas heterocigotas u homocigotas para la PCT pueden estar predispuestas al desarrollo de la enfermedad.
Diagnóstico
Las lesiones crónicas ampollosas y costráceas sobre las áreas expuestas al sol son características de la PCT. La biopsia cutánea apoya el diagnóstico de la PCT, pero no es específica. Otras porfirias (en especial la porfiria variegata) pueden causar lesiones idénticas. Ciertos fármacos y agentes fotosensibilizantes desconocidos pueden causar una seudoporfiria en la cual las lesiones se parecen a las de la PCT, pero con un aumento de las porfirinas.
El análisis de las porfirinas es esencial para el diagnóstico: todas las porfirias que causan lesiones cutáneas tendrán unas porfirinas plasmáticas elevadas. En la PCT, las porfirinas aumentan también en la orina y en menor medida en las heces con un patrón característico. Las porfirinas en la orina son en su mayor parte uroporfirinas y porfirinas heptacarboxílicas, con aumentos menores de la coproporfirina y de las porfirinas con 5 y 6 carboxilos. En el suero o en la orina pueden detectarse pequeñas cantidades de isocoproporfirinas, pero en las heces éstas suelen ser las porfirinas dominantes en la excreción; el aumento de la isocoproporfirina fecal (o un aumento del cociente isocoproporfirina/coproporfirina) es virtualmente diagnóstico de PCT. Esas infrecuentes porfirinas tetracarboxílicas se originan cuando se acumula porfirinógeno pentacarboxílico a causa de la deficiencia de la uroporfirinógeno descarboxilasa y después es metabolizado parcialmente por la coproporfirinógeno oxidasa para producir isocoproporfirinógeno.
La PCT tipo II se detecta mediante el hallazgo de uroporfirinógeno descarboxilasa disminuida en los eritrocitos junto con el patrón de exceso de porfirinas característico de la PCT. Esta enzima debe determinarse antes de practicar una flebotomía, porque el aumento de la eritropoyesis puede aumentar la enzima en los eritrocitos y hacer más difícil la detección de una deficiencia. El hallazgo de una cantidad reducida de la enzima no modifica el tratamiento. Se han descrito familias con herencia aparente de una PCT con una uroporfirinógeno descarboxilasa normal; esto se denomina a veces PCT tipo III, pero se desconoce la naturaleza del defecto genético.
Tratamiento
La PCT es la porfiria más fácil de tratar. El tratamiento inicial consiste en identificar los factores desencadenantes y evitarlos en la medida de lo posible, aunque el beneficio clínico obtenido es muy variable. La flebotomía suele ser eficaz en la inducción de remisiones clínicas, y es el tratamiento más ampliamente recomendado. Se extraen aproximadamente 400 ml de sangre cada 1 a 2 sem, y generalmente sólo se necesitan cinco o seis flebotomías. Esta medida depleciona el hígado de hierro haciendo al Paciente ligeramente deficiente en hierro. La flebotomía se interrumpe cuando el nivel de ferritina sérica (una medida de los depósitos de hierro corporal) desciende un poco por debajo de la normalidad. Una flebotomía excesiva puede causar anemia. Existen pruebas fehacientes de que los beneficios de la flebotomía son consecuencia de la disminución de los depósitos de hierro en el organismo. Las porfirinas urinarias y plasmáticas disminuyen gradualmente con el tratamiento, retrasándose, pero en forma paralela respecto al descenso de la ferritina. La piel mejora y con el tiempo se normaliza. Tras la remisión es innecesario continuar el tratamiento o mantener un nivel de ferritina bajo. Sólo es necesaria una nueva flebotomía si hay una recurrencia. La abstinencia de alcohol ayuda a mantener la remisión. Un tratamiento con estrógenos puede reanudarse tras la flebotomía cuando puede beneficiar la salud, como en las mujeres posmenopáusicas, y no suele causar una recurrencia.
Cuando la flebotomía no es factible, una opción útil es la cloroquina o la hidroxicloroquina en dosis de la mitad de una tableta estándar correspondiente a 125 o 100 mg v.o., respectivamente, dos veces a la semana. Estos fármacos eliminan el exceso de porfirinas del hígado. Las dosis más altas eliminan las porfirinas con demasiada rapidez, causando un empeoramiento transitorio de la porfiria y lesión del hígado. La pauta de dosis bajas puede interrumpirse después de lograrse la remisión. Es probable que la eficacia del tratamiento con cloroquina y con flebotomía sea parecida.
Tanto la PCT familiar como la no familiar responden a la flebotomía y a la cloroquina a dosis bajas, pero no otros tipos de porfiria. Por consiguiente, es importante un diagnóstico exacto antes de iniciar el tratamiento.
El diagnóstico y el tratamiento de la PCT son difíciles en Pacientes con una nefropatía terminal concurrente. Los niveles plasmáticos de porfirinas suelen ser mucho más elevados porque es escasa o no existe la excreción renal de las porfirinas, y éstas dializan mal. Dada la gran elevación de los niveles de porfirinas, las lesiones cutáneas pueden ser mucho más graves. El diagnóstico se establece principalmente midiendo las porfirinas plasmáticas y fecales. La flebotomía suele estar contraindicada a causa de la anemia (debida generalmente a deficiencia de eritropoyetina), y la cloroquina y la hidroxicloroquina no son eficaces. El tratamiento de reposición con eritropoyetina puede estimular la eritropoyesis, movilizar el exceso de hierro, apoyar la flebotomía y provocar la remisión de la PCT en estos Pacientes.
PROTOPORFIRIA ERITROPOYÉTICA
(Protoporfiria; protoporfiria eritrohepática)
Trastorno dominante autosómico, la forma más frecuente de porfiria eritropoyética y probablemente la tercera forma más frecuente de porfiria, debida a una deficiencia de ferroquelatasa.
En la protoporfiria eritropoyética (PPE), una deficiencia de ferroquelatasa (v. fig. 14-1, enzima 8) origina un agregado de porfirinas en la médula ósea y los eritrocitos. Este agregado pasa al plasma y es excretado por el hígado hacia la bilis y las heces. La enfermedad se caracteriza por la aparición de fotosensibilidad cutánea en la infancia, que se manifiesta principalmente por dolor, enrojecimiento y tumefacción inmediatamente después de la exposición a la luz solar. La prevalencia de la PPE no ha sido calculada de un modo fiable. No existe predilección racial o sexual. Una forma bovina de PPE es autosómica recesiva.
Se han encontrado muchas mutaciones diferentes en el gen de la ferroquelatasa en diferentes familias con PPE. La gravedad de la PPE varía considerablemente de unos Pacientes a otros; esta variación se produce incluso dentro de una misma familia en la cual muchas personas heredan la misma mutación, pero tienen poco o ningún aumento de la protoporfirina en los eritrocitos. Por consiguiente, la variación en la gravedad de la PPE no se explica del todo por la naturaleza de las diferentes mutaciones. En lugar de ello, puede deberse a un rasgo genético que conduce a una expresión del gen de la ferroquelatasa normal heredado a partir del otro progenitor. En consecuencia, los Pacientes con manifestaciones clínicas de PPE pueden tener actividades de ferroquelatasa inferiores a la mitad de la actividad normal que sería de esperar simplemente por la herencia autosómica dominante.
Síntomas y signos
Los síntomas suelen iniciarse al principio de la vida. Las sensaciones de quemazón dolorosa, el prurito, el eritema y la tumefacción que pueden parecerse al angioedema aparecen en seguida tras la exposición a la luz solar (a veces en el curso de minutos). Estos síntomas no son característicos de otras porfirias. La piel correosa y engrosada en el dorso de las manos, la leve cicatrización y los cambios en las uñas pueden producirse con la frecuente exposición al sol, pero las ampollas y las cicatrices graves son infrecuentes. Dado que son mayores las molestias que el daño objetivo de la piel, la enfermedad puede pasar sin ser reconocida incluso en Pacientes con síntomas intensos. Los Pacientes pueden desarrollar cálculos biliares que contienen protoporfirina, lo que refleja la alta concentración de la protoporfirina en la bilis.
La enfermedad puede presentarse también con anomalías leves, sin explicación, detectadas en las pruebas de la función hepática. Una hepatopatía crónica, aunque infrecuente, puede ser grave. La insuficiencia hepática se produce pocas veces, pero puede progresar con rapidez. La disfunción hepática progresiva en la PPE se asocia con concentraciones crecientes de protoporfirina en hígado, plasma y eritrocitos, y un empeoramiento de la fotosensibilidad. El daño hepático se debe en parte a los efectos tóxicos y colestásicos de las grandes cantidades de protoporfirina sobre el hígado. Si se presenta también una afección hepática simultánea, la insuficiencia hepática puede ser reversible. De lo contrario, el tratamiento suele ser ineficaz y puede ser imprescindible un trasplante de hígado. Las graves lesiones por fotosensibilidad se parecen a quemaduras (especialmente tras la exposición a las luces del quirófano) e incluso puede producirse la neuropatía en la hepatopatía protoporfírica avanzada.
Diagnóstico
Debe sospecharse una PPE en los Pacientes que se quejan de fotosensibilidad cutánea de aparición temprana en la vida, pero que no tienen ampollas ni formación de cicatrices. Es frecuente una historia familiar negativa. Las concentraciones de protoporfirinas en eritrocitos y plasma están notablemente aumentadas, pero no las porfirinas urinarias. El aumento de las protoporfirinas eritrocitarias es inespecífico; puede producirse en la deficiencia de hierro, en la intoxicación saturnina, en muchos trastornos eritrocitarios, en todas las porfirias recesivas autosómicas y a veces en las porfirias agudas dominantes autosómicas. Por lo demás, el aumento de los niveles plasmáticos de porfirinas sólo ocurre rara vez en patologías distintas de las porfirias que causan sensibilidad cutánea.
En todos los trastornos distintos de la PPE en los cuales las protoporfirinas eritrocitarias están aumentadas, incluidas algunas otras porfirias, el exceso de protoporfirina en las células rojas está formando un complejo con el cinc, mientras que en la PPE la protoporfirina está exenta de cinc. Las protoporfirinas-cinc y las protoporfirinas exentas de cinc no se miden por separado en la mayoría de los laboratorios. Dado que se dice a veces (incorrectamente) que la intoxicación por plomo está asociada con aumentos de la protoporfirina eritrocitaria libre, puede no estar claro si un informe de la concentración de protoporfirina eritrocitaria libre se refiere a una protoporfirina exenta de metal o a protoporfirina que forma parte de un complejo con el cinc. En la PPE pueden producirse aumentos notables de las protoporfirinas en las heces. Determinar las protoporfirinas en los eritrocitos, el plasma y las heces puede ser de utilidad en la detección selectiva de la enfermedad en los familiares de los Pacientes. La detección de una mutación hereditaria de la ferroquelatasa en los familiares puede ser factible si se ha identificado la mutación exacta en un caso índice.
Tratamiento y pronóstico
La fotosensibilidad se trata evitando la luz solar. El b-caroteno, cuando se toma en cantidades suficientes para causar un tono ligeramente amarillento de la piel, es especialmente eficaz para tratar esta porfiria; 120 a 180 mg/d v.o. mejoran la tolerancia a la luz solar en muchos Pacientes. El nivel sérico recomendado de b-caroteno es de 600 a 800 mg/dl; los efectos beneficiosos aparecen normalmente 1 a 3 meses después de comenzar el tratamiento. La protoporfirina excretada en la bilis puede ser reabsorbida en parte por el intestino y ser devuelta, a través de la sangre, hacia el hígado. Se han administrado con cierto éxito resinas y otros agentes fijadores para interrumpir esta circulación enterohepática. Los fármacos nocivos en las porfirias hepáticas no parecen empeorar las porfirias eritropoyéticas, pero se evitan por precaución. La deficiencia de hierro podría contribuir a un deterioro de la actividad de la ferroquelatasa y debe ser tratada.
A diferencia de las porfirias hepáticas, la evolución de la PPE es estable a lo largo del tiempo, y existen pocos cambios en los niveles de protoporfirina en el plasma y los eritrocitos, salvo si se presentan complicaciones hepáticas. El tratamiento de la insuficiencia hepática protoporfírica es complicado y puede requerir un trasplante de hígado.
PORFIRIAS MENOS FRECUENTES
DEFICIENCIA DE ÁCIDO DELTA-AMINOLEVULÍNICO DESHIDRATASA
(Porfiria deficiente en ALA-deshidratasa)
Trastorno recesivo autosómico, la porfiria más rara, que aparece por una deficiencia de ALA-deshidratasa.
Se han descrito varias mutaciones diferentes en el gen de la ALA-deshidratasa (v. fig. 14-1, enzima 2) en Pacientes no emparentados. La enfermedad fue descrita por primera vez en Alemania, pero probablemente se produce en todos los países. Causa síntomas neurológicos y a veces anemia.
Los síntomas y signos se parecen a los de las porfirias agudas, pero también incluyen hemólisis y anemia. Los síntomas pueden iniciarse en la lactancia o en la edad adulta. El ALA y la coproporfirina III urinarios y la protoporfirina-cinc eritrocitaria están notablemente aumentados. En éste y otros trastornos en los que se acumula ALA, su exceso puede metabolizarse a coproporfirina III en tejidos distintos del tejido de origen del exceso de ALA. La excreción fecal de porfirinas está normal o escasamente elevada. Los Pacientes con porfiria por deficiencia de ALA-deshidratasa (PAD) muestran poca actividad de ALA-deshidratasa en los eritrocitos o en células no eritroides, mientras que sus padres presentan alrededor de un 50% de la actividad enzimática.
El diagnóstico se hace con el hallazgo de un exceso de ALA y coproporfirinas en la orina y una deficiencia de ALA-deshidratasa en los eritrocitos. Hay que descartar otras causas de esta deficiencia enzimática, como la intoxicación por el plomo y la tirosinemia. Estos trastornos también pueden presentar síntomas (dolor abdominal, íleo y neuropatía motora) que son sorprendentemente similares a los de las porfirias agudas. En la intoxicación por plomo, la deficiencia de ALA-deshidratasa puede restaurarse a la normalidad in vitro con el ditiotreitol, un reactivo de grupos sulfhidrilos, mientras que una deficiencia genética de esta enzima no se restaura. En la tirosinemia hereditaria, una deficiencia congénita de la fumarilacetoacetasa conduce a la acumulación de succinilacetona (ácido 2,3-dioxoheptanoico). Este análogo estructural del ALA es un potente inhibidor de la deshidratasa. Otros metales pesados o la exposición al estireno también pueden inhibir la ALA-deshidratasa.
La experiencia con el tratamiento de la PAD es escasa, pero el tratamiento puede abordarse como el de una porfiria intermitente aguda.
PORFIRIA ERITROPOYÉTICA CONGÉNITA
(Enfermedad de Günther; porfiria eritropoyética; porfiria congénita; hematoporfiria congénita; uroporfiria eritropoyética)
Enfermedad autosómica recesiva, rara y generalmente grave, producida por una deficiencia de la uroporfirinógeno III cosintetasa.
Se han comunicado menos de 200 casos de porfiria eritropoyética congénita (PEC). No hay un claro predominio racial o sexual. Esta porfiria se produce raras veces en animales (ganado vacuno). Curiosamente, todas las ardillas zorrunas tienen esta enfermedad, la cual no parece afectarles desfavorablemente, aun cuando las porfirinas están notablemente aumentadas.
Patogenia
Se han identificado muchas mutaciones diferentes del gen de la uroporfirinógeno III cosintetasa humana (v. fig. 14-1, enzima 4). La mayoría de los Pacientes tienen padres no emparentados y han heredado una mutación diferente de cada uno de ellos. La actividad de la cosintetasa residual persiste incluso en los casos más graves. De hecho la producción de hemo aumenta en respuesta a una anemia hemolítica, pero esto tiene lugar a expensas de una considerable acumulación de hidroximetilbilano (HMB), el substrato de la enzima deficiente. El exceso de HMB es convertido (ciclado) no enzimáticamente a uroporfirinógeno I y después enzimáticamente a coproporfirinógeno I. Los porfirinógenos de tipo I no son precursores del hemo, y cuando se acumulan se oxidan espontáneamente a las correspondientes porfirinas. El exceso de porfirinas se acumula en la médula ósea, sobre todo en la etapa de maduración de la célula eritroide, cuando la síntesis de hemoglobina es más activa. Las porfirinas causan hemólisis intramedular y también acortan la supervivencia de los eritrocitos circulantes.
Síntomas y signos
La formación de ampollas cutáneas suele ser intensa, empieza pronto después del nacimiento y se acompaña de anemia y orina de color rojo. Algunos casos relativamente leves comienzan en la vida adulta. La gravedad depende de las mutaciones halladas en cada Paciente y del grado de deficiencia de la enzima.
Muchos casos graves que se han presentado en forma de hidropesía no inmunológica in utero requieren transfusiones intravenosas y (si la enfermedad no fue identificada en seguida tras el nacimiento) apareció una intensa fotosensibilidad cuando se inició la fototerapia para la ictericia neonatal. Este tipo de casos pueden beneficiarse del diagnóstico in utero. El líquido amniótico puede ser de color rojizo por el alto contenido de porfirinas.
Los cambios cutáneos son similares a los de la PCT, pero en general son más graves. Las lesiones ampollosas de la piel expuesta al sol suelen conducir a la formación de cicatrices, a infección y a pérdida de rasgos faciales y de dedos. Son comunes las alteraciones de la pigmentación y la hipertricosis. La formación de cicatrices en la córnea puede ser grave. Las porfirinas se depositan en los dientes (produciendo una coloración pardo-rojiza denominada eritrodoncia) y en los huesos. La desmineralización ósea puede ser considerable. Casi siempre se presenta anemia hemolítica y esplenomegalia. Esta última puede contribuir a la anemia y causar leucopenia y trombocitopenia (hiperesplenismo). La anemia estimula a la médula ósea a generar más células eritroides cargadas de porfirinas, y con ello a una producción de porfirinas creciente y a la perpetuación de la hemólisis y la fotosensibilidad. Los fármacos, las hormonas (distintas de la eritropoyetina endógena) y la nutrición (distinta de las deficiencias vitamínicas que pueden deteriorar la médula ósea) tienen poca influencia sobre la enfermedad. No hay manifestaciones neurológicas.
Diagnóstico
El diagnóstico de la PEC lo sugiere la presencia de orina de color rosado a pardo oscuro o la aparición de una intensa fotosensibilidad en la lactancia (o, pocas veces, en los adultos). También puede presentarse in utero en forma de hídrops de origen no inmunológico. Las porfirinas están aumentadas en la médula ósea, los eritrocitos, en el plasma y la orina con un patrón característico, generalmente con niveles mucho más altos que los de otras porfirinas. La uroporfirina I y la coproporfirina I son las porfirinas predominantes en orina, plasma y eritrocitos, y la coproporfirina I es la porfirina predominante en las heces. Los eritrocitos contienen a veces grandes cantidades de protoporfirinas, como en otras porfirias homocigotas. La demostración de una deficiencia de actividad de la uroporfirinógeno cosintetasa confirma el diagnóstico. El ALA y el PBG no están aumentados. La porfiria hematoeritropoyética es clínicamente similar, pero los patrones de porfirinas son diferentes. Las manifestaciones cutáneas y los patrones de porfirinas en la protoporfiria eritropoyética son distintos de los de la PEC y otras porfirias.
Tratamiento y prevención
El tratamiento no es muy eficaz. Por tanto, es importante evitar la luz solar y llevar ropa protectora. (Varios fabricantes se especializan en telas y vestidos protectores para personas sensibles al sol.) Evitar los traumatismos cutáneos menores y tratar inmediatamente las infecciones bacterianas secundarias ayuda a prevenir la cicatrización y el desfiguramiento. La esplenectomía puede mejorar la anemia hemolítica. Las transfusiones de eritrocitos para corregir la anemia pueden reducir la producción de porfirinas. El trasplante de médula ósea y, en el futuro, el tratamiento genético son otras opciones. En las familias afectadas se pueden detectar los individuos heterocigotos, y el diagnóstico in utero es posible. Por consiguiente, existen opciones para la prevención de la transmisión genética.
PORFIRIA HEPATOERITROPOYÉTICA
Enfermedad autosómica recesiva muy rara, generalmente grave, producida por una deficiencia de uroporfirinógeno descarboxilasa.
La deficiencia de uroporfirinógeno descarboxilasa (v. fig. 14-1, enzima 5) está presente en todos los tejidos y se determina de manera más práctica en los eritrocitos. Aunque el grado de la deficiencia es intenso, persiste alguna actividad enzimática residual. El trastorno es menos grave en los casos con más actividad enzimática residual.
Se han comunicado en todo el mundo menos de 20 casos de porfiria hepatoeritropoyética (PHE). La formación de ampollas en la piel, la orina roja y la anemia son frecuentes. Aunque la PHE es clínicamente muy similar a la porfiria eritropoyética congénita, estas enfermedades difieren en sus patrones de acumulación de porfirinas.
El patrón de acumulación de porfirinas de la PHE se parece al de la porfiria cutánea tarda, excepto en que la protoporfirina-cinc eritrocitaria también está aumentada. Los rasgos diagnósticos son isocoproporfirina fecal o urinaria y protoporfirina-cinc eritrocitaria elevadas. La flebotomía puede ser beneficiosa en los casos de PHE más leves. El tratamiento de los casos más graves es similar al de la porfiria eritropoyética congénita.
COPROPORFIRIA HEREDITARIA
Enfermedad autosómica dominante producida por una deficiencia de coproporfirinógeno oxidasa.
La coproporfiria hereditaria (CPH) es similar a la porfiria intermitente aguda, aunque es menos frecuente, y a menudo más leve, y a veces se asocia con fotosensibilidad. Se han descrito unos pocos casos homocigotos.
Como otras porfirias, la CPH es genéticamente heterogénea en el nivel del gen de la coproporfirinógeno oxidasa (v. fig. 14-1, enzima 6). Esta enzima cataliza la descarboxilación en dos pasos del coproporfirinógeno III a protoporfirinógeno IX. Un producto intermedio es un porfirinógeno tricarboxílico denominado harderoporfirinógeno. En una variante bioquímica de la CPH, llamada harderoporfiria, una mutación causa una alteración estructural en la enzima que reduce la afinidad para el substrato, y se acumula harderoporfirina y también coproporfirina.
Síntomas y signos
Los ataques agudos de síntomas abdominales y neurológicos son desencadenados por los mismos factores que son importantes en la porfiria intermitente aguda, como ciertos fármacos (p. ej., barbituratos, sulfonamidas) y esteroides (especialmente progesterona). A veces se produce fotosensibilidad, pero con menos frecuencia que en la porfiria variegata.
Diagnóstico y tratamiento
El diagnóstico se funda en el hallazgo de cantidades aumentadas de ALA, PBG y coproporfirinas en orina y exceso de coproporfirinas en heces. Una presencia predominante o exclusiva de coproporfirina fecal es más sugerente de CPH que de porfiria variegata (v. más adelante), en la cual las concentraciones fecales de coproporfirinas y protoporfirinas suelen ser aproximadamente iguales. La excreción urinaria de ALA, PBG y uroporfirinas puede estar aumentada durante los ataques agudos. La excreción se normaliza entre los ataques con más frecuencia que en la porfiria intermitente aguda. La deficiencia de coproporfirinógeno oxidasa puede demostrarse en células distintas de los eritrocitos, pero no está recomendada para el diagnóstico de rutina. El tratamiento de los ataques agudos es el de la porfiria intermitente aguda.
PORFIRIA VARIEGATA
(Porfiria variegata; protocoproporfiria; porfiria genética sudafricana)
Enfermedad autosómica dominante producida por una deficiencia de protoporfirinógeno oxidasa.
La porfiria variegata (PV) es prevalente en Sudáfrica, donde la mayoría de los casos se han rastreado hasta una pareja que inmigró desde Holanda en los últimos años del siglo xvii, uno de cuyos miembros era portador del rasgo. La mayoría de los Pacientes de PV en Sudáfrica son descendientes de esa persona, y por tanto tienen la misma mutación específica. La PV también existe en muchas otras razas. Los heterocigotos tienen aproximadamente un 50% de deficiencia de protoporfirinógeno oxidasa (v. fig. 14-1, enzima 7), y la mayoría nunca desarrollan síntomas. Se han descrito unos pocos casos con deficiencias homocigotas en la protoporfirinógeno oxidasa.
Esta enzima es la última de las de la vía del hemo que ha sido clonada y secuenciada, y se han identificado recientemente muchas mutaciones diferentes en familias no emparentadas. Como era de esperar, en Sudáfrica es especialmente frecuente una sola mutación.
Síntomas y signos
Los síntomas y signos de la PV son los mismos que los de la porfiria intermitente aguda, a excepción de que algunos Pacientes presentan fotosensibilidad. Las lesiones cutáneas son indistinguibles de las de la porfiria cutánea tarda. Los mismos factores que son perjudiciales en otras porfirias agudas pueden provocar ataques de PV. Además de los síntomas neuroviscerales, suelen producirse manifestaciones cutáneas, y se presentan con menor frecuencia en los climas fríos que en los cálidos, donde la luz solar es más intensa.
Los niveles de ALA y PBG están aumentados, especialmente durante los ataques agudos. Este aumento refleja la inducción de la ALA-sintasa hepática por factores como esteroides endógenos, fármacos y alteraciones nutricionales. Cuando ocurre esto, la PBG-desaminasa, cuya actividad en el hígado es normalmente tan baja como la de la ALA-sintasa, puede convertirse en el paso limitante de la velocidad de la vía en tal forma que el PBG se acumula. En este trastorno, el exceso de protoporfirinógeno en el hígado puede inhibir a la PBG-desaminasa. La acumulación de coproporfirinógeno es comprensible por la asociación funcional que existe entre la coproporfirinógeno oxidasa y la protoporfirinógeno oxidasa en las mitocondrias. Además, el coproporfirinógeno desaparece de las células hepáticas más fácilmente que otros porfirinógenos, y sus pérdidas aumentan cuando la vía del hemo está estimulada.
Diagnóstico
La PV debe tenerse en cuenta en el diagnóstico diferencial de las porfirias agudas, especialmente si la actividad de la PBG-desaminasa es normal. Como en el caso de la coproporfiria hereditaria, los precursores de las porfirinas urinarias y las uroporfirinas están aumentados durante los ataques agudos. Cuando un ataque remite, aquéllos se normalizan más fácilmente que en la porfiria intermitente aguda. En la PV, las coproporfirinas urinarias están notablemente aumentadas y por lo general de modo persistente. Mientras que un aumento aislado intenso de la coproporfirina fecal es un rasgo distintivo de la coproporfiria hereditaria, en la PV las coproporfirinas y las protoporfirinas están aumentadas aproximadamente en la misma medida. El espectro de fluorescencia de las porfirinas plasmáticas (tras la dilución del plasma a pH neutro) es característico y muy útil para distinguir rápidamente la PV de las demás porfirias. (El máximo de emisión refleja probablemente los conjugados porfirina-péptido característicos.) En los adultos, incluso en los casos latentes, esta prueba es el método más sensible para detectar esta enfermedad.
Tratamiento
El tratamiento de los ataques agudos es el mismo que para los de la porfiria intermitente aguda. Son útiles las medidas que protegen la piel de la luz solar. A veces la colestiramina puede reducir la fotosensibilidad. Las flebotomías y la cloroquina no son eficaces.
PORFIRIA DUAL
Enfermedades producidas por deficiencias de más de una enzima de la vía biosintética del hemo.
Aunque las mutaciones de las enzimas de la vía biosintética del hemo son infrecuentes, a veces una única persona puede heredar una deficiencia en más de una de esas enzimas. Por ejemplo, se ha descrito el caso de Pacientes con deficiencias de uroporfirinógeno descarboxilasa y también de protoporfirinógeno oxidasa. Las personas que tienen deficiencias hereditarias de PBG-desaminasa y de uroporfirinógeno descarboxilasa pueden tener síntomas de porfiria aguda, porfiria cutánea, o de ambas. También se han descrito porfirias duales debidas a deficiencias de coproporfirinógeno oxidasa y de uroporfirinógeno III cosintetasa, así como de PBG-desaminasa y de coproporfirinógeno desaminasa.