224 / ENFERMEDADES GLOMERULARES
(Glomerulopatías)
Diversos procesos patológicos que afectan fundamentalmente al glomérulo e incluyen a las glomerulonefritis, aunque no se limitan a ellas.
Las enfermedades glomerulares pueden ser primarias o secundarias a enfermedades sistémicas. Entre las principales categorías patogénicas destacan la inflamatoria (síndrome nefrítico) y la hemodinámica (síndrome nefrótico). Se emplea el término síndrome nefrítico agudo como sinónimo de glomerulonefritis aguda (GA), cuyo prototipo es la GA postestreptocócica. También existen enfermedades nefríticas crónicas (síndrome nefrítico-proteinúrico crónico). El síndrome nefrótico es un conjunto de signos, síntomas y hallazgos de laboratorio debidos al aumento de la permeabilidad de la membrana capilar glomerular. Muchas enfermedades renales pueden provocar un síndrome nefrótico y se pueden asociar síndrome nefrótico y nefrítico.
Existen semejanzas estructurales, funcionales y clínicas dentro de cada grupo, porque el riñón responde ante una agresión de muy pocas formas y las enfermedades renales determinan escasos signos o síntomas. Aunque las diferencias morfológicas pueden ayudar a entender las glomerulopatías, las correlaciones clínico-morfológicas se consideran indicadores poco fiables del pronóstico y de la respuesta al tratamiento.
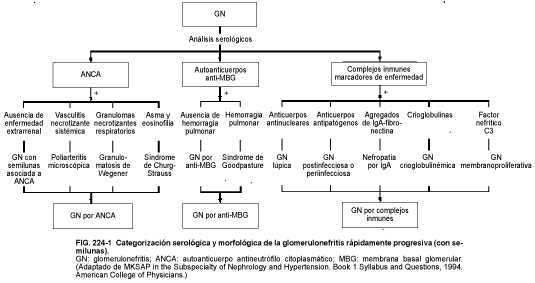
En algunos pacientes con glomerulopatías se producen manifestaciones extrarrenales que sugieren la enfermedad causal (vasculitis sistémicas, infecciones). Los marcadores serológicos, como los anticuerpos antiestreptocócicos, los anticuerpos antimembrana basal glomerular (MBG), los autoanticuerpos antineutrófilo citoplasmáticos (ANCA), los anticuerpos antinucleares, las crioglobulinas, el factor nefrítico C3, la hipocomplementemia o los agregados de IgA-fibronectina, sugieren una glomerulopatía específica o, al menos, permiten acotar el diagnóstico diferencial (v. fig. 224-1).
SÍNDROME NEFRÍTICO
El síndrome nefrítico clásico incluye hematuria, hipertensión, insuficiencia renal y edema, aunque con frecuencia faltan algunos elementos. El síndrome nefrítico puede ser agudo o transitorio (GA postinfecciosa), fulminante con una insuficiencia renal rápida (glomerulonefritis rápidamente progresiva [GNRP]) o indolente (p. ej., nefropatía IgA). Los cambios morfológicos y, con frecuencia, las manifestaciones clínicas varían a lo largo del tiempo.
SÍNDROME NEFRÍTICO AGUDO
(Glomerulonefritis aguda, glomerulonefritis post-infecciosa)
Síndrome caracterizado morfológicamente por cambios inflamatorios difusos en los glomérulos y clínicamente por la aparición súbita de hematuria con cilindros de hematíes, leve proteinuria y, con frecuencia, hipertensión, edema y azoemia.
Etiología
El prototipo de síndrome nefrítico agudo es la glomerulonefritis postestreptocócica (GNPE), debida a la infección por determinadas cepas nefritogénicas de estreptococos b-hemolíticos del grupo A, como la tipo 12 (asociada con faringitis) y la tipo 49 (asociada con impétigo). La incidencia de GNPE está disminuyendo en EE. UU. y Europa. En otras zonas del mundo donde se producen epidemias, de un 5 a un 10% de los pacientes con faringitis y aproximadamente el 25% de los pacientes con infecciones cutáneas desarrollan este tipo de glomerulonefritis. Se produce con más frecuencia en niños >3 años y adultos jóvenes, pero un 5% de los pacientes son mayores de 50 años. Se produce un período de latencia entre 1 y 6 sem (media, 2 sem) desde la infección hasta el inicio de los síntomas de GN.
Morfología y patogenia
Las lesiones se localizan principalmente en los glomérulos, que aumentan de tamaño y aparecen hipercelulares, primero por la presencia de neutrófilos y eosinófilos y luego por mononucleares. Se produce con frecuencia una hiperplasia transitoria de las células epiteliales en fases iniciales del proceso. Se pueden producir microtrombosis; si las lesiones son graves, los cambios hemodinámicos producen oliguria, con frecuente formación de semilunas epiteliales (formadas en el espacio de Bowman por la hiperplasia de las células epiteliales, mediada posiblemente por factores de crecimiento secretados por los macrófagos estimulados). Aumenta el número de células endoteliales y mesangiales y las regiones mesangiales suelen aparecer más expandidas por el edema y contienen neutrófilos, células muertas, restos celulares y depósitos subepiteliales de material electrodenso.
El estudio con microscopio de fluorescencia suele demostrar depósitos de complejos inmunes con IgG y complemento en un patrón granular. Con microscopio electrónico estos depósitos adoptan una morfología de semilunas o jorobas y se localizan en la zona subepitelial. La presencia de estos depósitos indica una reacción inflamatoria mediada por complemento (descrita en cap. 231), que determina la lesión glomerular. Aunque se considera que el complejo inmune contiene un antígeno relacionado con gérmenes estreptocócicos, no se ha conseguido identificar dicho antígeno.
Signos y síntomas
Las manifestaciones iniciales van desde una hematuria asintomática (un 50% de casos) y proteinuria leve a una nefritis bien desarrollada con hematuria macroscópica o microscópica (de color coca-cola, parda, oscura o francamente hemática), proteinuria, oliguria, edema, hipertensión e insuficiencia renal.
En un 10% de los adultos y un 1% de los niños el síndrome nefrítico agudo evoluciona a una GNRP. En los pacientes con enfermedad remitente, la proliferación celular desaparece en semanas, pero la intensidad de la respuesta inflamatoria varía mucho, siendo frecuente la esclerosis residual. La mayor parte de los niños (85 a 95%) conservan o recuperan la función renal normal, sobre todo si adquieren la enfermedad durante una infección estreptocócica. En pocas ocasiones, sobre todo esporádicas y en adultos, sólo se produce una recuperación parcial, pudiendo persistir la hematuria o la proteinuria, detectables analíticamente durante años.
Hallazgos de laboratorio y diagnóstico
Se pueden excretar >0,5 a 2 g/m2/d de proteínas; el cociente proteínas/creatinina en orina puede ser <2 (normal 0,1 a 0,3). En el sedimento urinario se reconocen hematíes dismórficos, leucocitos y células tubulares renales, siendo característicos los cilindros que contienen hematíes y Hb, y frecuentes los que contienen polinucleares y los cilindros granulosos (gotas de proteínas).
El título de anticuerpos contra el agente infeccioso causante suele aumentar en 1 a 2 sem. Se puede medir el incremento de los anticuerpos contra los productos antigénicos de los estreptococos: la antiestreptolisina O (ASO) es el mejor indicador de infecciones de vías respiratorias altas, y antihialuronidasa y antideoxiribonucleasa B lo son del pioderma. C3 y C4 suelen ser bajos durante la enfermedad activa (v. tabla 224-1), pero los niveles de complemento se suelen normalizar en 6 a 8 sem en un 80% de los casos de GNPE, aunque no lo hacen prácticamente en ningún caso de glomerulonefritis membranoproliferativa (GNMP). La crioglobulinemia suele persistir durante varios meses, a diferencia de los complejos inmunes circulantes, que sólo se pueden detectar algunas semanas.

La función tubular se suele alterar por los cambios inflamatorios intersticiales, reduciendo la capacidad de concentración urinaria y la excreción de ácido y determinando diversas alteraciones en el intercambio de solutos en la nefrona. Como existe cierta capacidad intrínseca para la hipertrofia glomerular, dichos defectos de la función tubular se suelen observar antes de que el IFG se reduzca mucho. Al progresar las alteraciones glomerulares, se reduce de forma significativa la superficie total de filtración, con el consiguiente descenso del IFG y el desarrollo de azoemia. Se puede calcular el IFG a partir de la concentración de creatinina sérica o del aclaramiento urinario de creatinina. Aunque el IFG se suele normalizar en 1 a 3 meses, la proteinuria puede persistir durante 6 a 12 meses y la hematuria microscópica durante varios años. Los cambios en el sedimento urinario pueden reaparecer con infecciones respiratorias altas leves.
Los antecedentes de dolor de garganta, impétigo o infección estreptocócica demostrada con cultivo 1 a 6 sem antes del inicio del síndrome y la elevación de los anticuerpos anti-estreptocócicos permiten realizar el diagnóstico. Los cilindros hemáticos son patognomónicos de cualquier GN, pero valorados en el contexto clínico se consideran altamente indicativos de síndrome nefrítico agudo. La ecografía permite distinguir la enfermedad aguda (los riñones suelen ser normales o algo grandes) de la exacerbación de una nefropatía crónica (riñones pequeños).
Pronóstico
El pronóstico depende de la edad del paciente, de si la enfermedad es esporádica o epidémica y del estadio de las lesiones renales cuando remite el estímulo inflamatorio. El pronóstico suele ser bueno cuando la lesión renal inicial no es grave y se puede reducir o eliminar la fuente de antigenemia. En la mayor parte de los pacientes se produce una desaparición gradual de los síntomas y los signos. En los casos graves se puede producir hipertensión con insuficiencia cardíaca y encefalopatía hipertensiva o sin ella. Un descenso importante del IFG o el desarrollo de un síndrome nefrótico (en un 30% de los pacientes, sobre todo en los que presentan muchos depósitos subepiteliales) con extensa formación de semilunas y necrosis, indica una rápida progresión a insuficiencia renal terminal. En pocos pacientes, el inicio de la enfermedad se acompaña de anuria, hipervolemia grave e hiperpotasemia, produciéndose la muerte si no se realiza diálisis.

Tratamiento
El tratamiento antibiótico de las infecciones antes de que se desarrolle una GNPE no parece prevenir este proceso. Si en el momento del diagnóstico existe una infección bacteriana, se debe iniciar el tratamiento antibiótico y tratar todas las causas secundarias (v. tablas 224-2 y 224-3). Los fármacos inmunosupresores resultan ineficaces y los esteroides pueden agravar el cuadro. Si se produce azoemia y acidosis metabólica, se debe restringir la ingesta proteica. La ingesta de Na sólo se reduce cuando existe sobrecarga circulatoria, edema o hipertensión grave; los diuréticos (tiazidas, diuréticos del asa) pueden ayudar a mantener el volumen del LEC expandido. La hipertensión exige un tratamiento enérgico (v. cap. 199). Los casos de insuficiencia renal grave necesitan diálisis (v. cap. 223).

Causas no bacterianas de síndrome nefrítico agudo
También se puede producir un síndrome nefrítico agudo tras las infecciones virales, parasitarias o micóticas. Las manifestaciones clínicas y morfológicas de estas formas pueden parecerse a las de la GNPE (sin presencia de «jorobas» subepiteliales) o la GNMP de tipo I (sin depósitos subendoteliales ni mesangiales). Se pueden producir muchos tipos de afectación renal y las manifestaciones sistémicas suelen recordar a las de otras enfermedades (poliarteritis nodosa, émbolos renales, nefritis intersticial aguda inducida por fármacos). Se puede observar una GN lúpica, aunque es más frecuente en caso de síndrome nefrótico (v. más adelante).
En general resulta más sencillo diagnosticar estas formas de glomerulonefritis que la GNPE, porque su período de latencia es más corto y se producen mientras la infección sigue siendo aparente. Sin embargo, puede resultar difícil diagnosticar la nefritis por VEB, sobre todo si los hemocultivos son negativos.
La gravedad de las manifestaciones clínicas depende de la duración de la infección; por ejemplo, el síndrome nefrítico agudo secundario a la infección de una prótesis vascular tiene buen pronóstico si se consigue erradicar dicha infección (en general asociada con Staphyloccus epidermidis), algo que suele exigir la retirada de la prótesis y antibioterapia. Sin embargo, se puede producir una insuficiencia renal irreversible en los pacientes con antecedentes de patología renal previa y en aquellos en los que se ha retrasado el tratamiento o que presentan lesiones extensas (semilunas, necrosis).
GLOMERULONEFRITIS RÁPIDAMENTE PROGRESIVAS
(Glomerulonefritis con semilunas)
Síndrome que se caracteriza morfológicamente por necrosis focal y segmentaria y proliferación de células epiteliales (semilunas) en la mayor parte de los glomérulos y clínicamente por una insuficiencia renal fulminante con proteinuria, hematuria y cilindros hemáticos.
Etiología, incidencia y clasificación
La glomerulonefritis rápidamente progresiva (GNRP) es infrecuente. Se suele ignorar su patogenia y las causas inmediatas son diversas (v. tabla 224-4 y fig. 224-1). Los estudios de inmunofluorescencia y biopsia permiten clasificar este proceso, aunque en algunos casos los datos de laboratorio sugieran más de un síndrome.

La GNRP pauciinmune representa el 50% de todos los casos de GNRP y se caracteriza por la ausencia de depósitos de complemento o complejos inmunes en el glomérulo. Los ANCA se consideran un marcador serológico de GNRP pauciinmune asociada con vasculitis sistémica o con los casos en que falta de evidencia de enfermedad extrarrenal. Los pacientes tienen anticuerpos contra la proteinasa 3 de los leucocitos (citoplasmática o C-ANCA), anticuerpos antimieloperoxidasa (perinucleares o P-ANCA) o contra ambos.
La GNRP por complejos inmunes representa un 40% de los casos de GNRP y parece que es idiopática, aunque se ha relacionado con la penicilamina, la sífilis y los tumores malignos en algunos pacientes. Suelen existir evidencias de enfermedad sistémica, como LES, púrpura de Henoch-Schönlein y crioglobulinemia mixta. En algunos pacientes se superpone la GNRP con otras enfermedades renales primarias (GNMP, nefropatía IgA, nefropatía membranosa).
La enfermedad por anticuerpos contra la MBG constituye un 10% de los casos de GNRP y es idiopática. El antígeno primario es un componente del colágeno de tipo IV, aunque también las células T autorreactivas contribuyen al desarrollo de los anticuerpos contra la MBG en el glomérulo y alvéolos. Se reconocen anticuerpos contra la MBG en la sangre y se pueden detectar en la membrana basal glomerular mediante inmunofluorescencia. En el 60 al 90% de los pacientes el anticuerpo reacciona de forma cruzada con la membrana basal alveolar y produce una alveolitis con hemorragia pulmonar (v. cap. 77). La lesión renal con afectación pulmonar (gripe, exposición a hidrocarburos, tabaquismo crónico) sugiere una lesión pulmonar simultánea, que permite que los anticuerpos circulantes accedan a los alvéolos.
Morfología
Se produce una proliferación focal de las células epiteliales glomerulares, a veces entremezcladas con numerosos neutrófilos, que forma una masa celular en semiluna, que rellena el espacio de Bowman en el 50 al 100% de los glomérulos renales. El penacho glomerular suele aparecer hipocelular y se colapsa. No es infrecuente la necrosis del penacho o de la semiluna, que puede representar la alteración más frecuente. En dichos pacientes se deben buscar evidencias de vasculitis.
El edema intersticial suele ser una característica notable en fases iniciales. Es difuso y se acompaña de infiltración por diversos tipos de células inflamatorias; cuando este fenómeno es extenso predominan los mononucleares. Entre los cambios iniciales en los túbulos destacan la formación de vacuolas y gotas hialinas y, en los túbulos distales, la formación de cilindros de hematíes y hialinos. Cuando progresa la enfermedad, se produce atrofia con engrosamiento de la MBG. El intersticio se fibrosa de forma difusa y disminuye el número de células inflamatorias.
La alteración más importante que se observa con microscopio de fluorescencia es el depósito lineal de IgG (generalmente asociado con C3 segmentario), aunque este patrón no es constante ni específico, pudiendo observarse en la nefropatía diabética y la GN fibrilar. Sin embargo, en estos casos el depósito de IgG es inespecífico y en semilunas, no se reconocen anticuerpos anti-MBG circulantes y existen otras alteraciones histológicas destacables (glomerulosclerosis diabética, fibrillas detectables con microscopía electrónica en la GN fibrilar).
En la GNRP por complejos inmunes grave se producen depósitos difusos irregulares de IgG y C3, con frecuente proliferación de las células intraglomerulares y formación de semilunas. En otros casos no resulta posible detectar depósitos de IgG o complemento. Sin embargo, se produce fibrina en las semilunas cualquiera que sea el patrón de fluorescencia.
Signos y síntomas
La presentación clínica puede ser similar a las enfermedades agudas no progresivas, pero el inicio suele ser más insidioso, destacando la debilidad, la fatiga y la fiebre. También se producen con frecuencia náuseas y vómitos, anorexia, artralgias y dolor abdominal. Un 50% de los pacientes presentan edema y antecedentes de un proceso seudogripal 4 sem antes del inicio de la insuficiencia renal, al que suele seguir una oliguria grave. Pocos pacientes tienen antecedentes de proteinuria. La hipertensión es poco frecuente y no suele ser grave. En algunas ocasiones se observa asociación entre la GNRP y manifestaciones pulmonares (síndrome reno-pulmonar, v. tabla 224-5).

Hallazgos de laboratorio y diagnóstico
Se caracteriza por una azoemia de gravedad variable. También existe siempre una hematuria, con frecuencia macroscópica, y cilindros de hematíes, siendo habitual el sedimento telescópico (cilindros leucocitarios, granulosos, céreos y anchos). La anemia, en ocasiones grave, es un hallazgo constante y la leucocitosis es frecuente.
Se debe sospechar una GNRP pauciinmune ante los pacientes con títulos de ANCA significativos. La granulomatosis de Wegener se asocia con C-ANCA en el 90% de los casos, mientras que la GN necrotizante idiopática se asocia con P-ANCA en el 80% de los casos. En la poliarteritis microscópica se observa una distribución relativamente igual de P-ANCA y C-ANCA.
La GNRP por complejos inmunes se debe sospechar ante un paciente con títulos elevados de anticuerpos frente a estreptococos, complejos inmunes circulantes o crioglobulinemia. La hipocomplementemia es frecuente en este tipo de GNRP, siendo poco habitual en la producida por anticuerpos anti-MBG. Resulta útil una serología positiva para anticuerpos anti-MBG circulantes, ya que los anticuerpos tienden a desaparecer en 3 a 6 meses. La ecografía o la radiografía (sin contraste) pueden mostrar unos riñones inicialmente aumentados de tamaño, que van disminuyendo de forma progresiva.
Resulta esencial obtener una biopsia en fases precoces de una posible GNRP para poder establecer el diagnóstico, determinar el pronóstico y planificar el tratamiento. También se deben realizar estudios serológicos y analizar los posibles procesos infecciosos.
Pronóstico y tratamiento
Si el síndrome es idiopático, la remisión espontánea es rara y un 80% de los pacientes evolucionan hacia una nefropatía terminal en 6 meses. Es frecuente la anuria irreversible y los pacientes no dializados mueren en pocas semanas. Sin embargo, la función renal puede mejorar con tratamiento en caso de GN postinfecciosa, lúpica, granulomatosis de Wegener o poliarteritis nodosa. Los pacientes que recuperan la función renal normal pueden tener cambios histológicos, sobre todo glomerulares, correspondientes fundamentalmente a hipercelularidad con poca o nula esclerosis en el penacho glomerular o en las células epiteliales y mínima fibrosis intersticial. El pronóstico es malo en los pacientes mayores de 60 años con insuficiencia renal oligúrica o en aquellos con semilunas circunferenciales en >75% de los glomérulos.
Cuando se observa una enfermedad por semilunas grave en una biopsia sin colapso extenso de los glomérulos, lesiones tubulointersticiales ni enfermedad o infección multisistémica, se debe iniciar precozmente el tratamiento (creatinina sérica <5 mg/dl [<440 m mol/l]), para que su eficacia sea óptima. Los pacientes sin enfermedad poranticuerpos anti-MBG que necesitan diálisis en fases iniciales pueden beneficiarse también deeste tratamiento. El tratamiento con pulsos de metilprednisolona (1 g/d i.v. en 30 min durante 3 a 5 d seguidos de 1 mg/kg/d de prednisona oral) puede llegar a reducir los niveles de creatinina sérica o retrasar la diálisis durante >3 años en un50% de los pacientes con GNRP pauciinmune o por complejos inmunes. Los pacientes con ANCA positivos se pueden beneficiar de la adición de ciclofosfamida oral, 1,5 a 2 mg/kg/d. La administración de pulsos mensuales de ciclofosfamida puede conseguir la reducción de los efectos adversos, pero todavía no se ha definido su importancia.
Cuando la respuesta al tratamiento farmacológico no es eficaz o es incompleta, se realiza una plasmaféresis para eliminar los anticuerpos libres, los complejos inmunes intactos y los mediadores de la inflamación (p. ej., fibrinógeno, complemento). La plasmaféresis con realización de intercambios de 4 litros diarios durante 14 d se considera el tratamiento de elección de la enfermedad por anticuerpos anti-MBG. En la GNRP pauciinmune o por complejos inmunes se suelen realizar intercambios de 3 a 4 litros durante 4 a 6d. La prednisona y la ciclofosfamida reducen al mínimo la formación de anticuerpos nuevos. Resulta fundamental la monitorización estrecha durante la plasmaféresis para evitar las infecciones que amenazan la vida y los efectos farmacológicos adversos.
La diálisis de mantenimiento debería ser sustituida por el tratamiento de la patología renal primaria. El trasplante renal presenta el riesgo de reaparición de la patología renal inicial sobre el riñón implantado.
SÍNDROME PROTEINÚRICO-HEMATÚRICO RENAL PRIMARIO
Conjunto de enfermedades que se suelen caracterizar por una hematuria macroscópica recidivante con proteinuria leve y cambios glomerulares.
La nefropatía por IgA, la enfermedad renal más importante, se produce en todas las edades, pero se observa con más frecuencia en niños y adultos jóvenes. Afecta a los varones 6 veces más que a las mujeres y es infrecuente en pacientes de raza negra. Su prevalencia respecto de las restantes enfermedades glomerulares primarias es del 5% en EE. UU., del 10 al 20% en Europa y Australia y del 30 al 40% en Asia. Otras enfermedades pueden cursar como un síndrome proteinúrico-hematúrico asintomático (v. tabla 224-6).

Patogenia
Un 50% de los pacientes con hematuria recidivante de origen renal presentan importantes depósitos mesangiales de IgA y la mitad de éstos tienen elevada la IgA sérica. También se han descrito alteraciones en las subpoblaciones de linfocitos T.
Las evidencias indican que la nefropatía por IgA se debe a una mayor producción o a un menor aclaramiento de complejos antigénicos poliméricos IgA, que se localizan en el mesangio y activan la vía clásica del complemento. Se supone que la IgA polimérica deriva de las superficies mucosas ricas en IgA.
Signos, síntomas y hallazgos de laboratorio
La forma de presentación más frecuente es una hematuria macroscópica recidivante o persistente (90% de los niños afectados) o una hematuria asintomática microscópica con una proteinuria leve (no nefrótica). La hematuria puede ser leve o grave, pero suele ser dismórfica. A veces se produce proteinuria sin hematuria. Aunque muchos pacientes no están realmente asintomáticos cuando se detecta por primera vez la alteración urinaria, pocos síntomas se pueden relacionar directamente con los riñones.
La función renal es normal en fases iniciales, pero se puede producir una patología renal sintomática. Pocos pacientes presentan una insuficiencia renal aguda o crónica, hipertensión grave o síndrome nefrótico (SN). El síndrome proteinúrico-hematúrico suele debutar 1 o 2 d después de una enfermedad febril mucosa (de vías respiratorias altas, sinusal o enteral), por lo que se parece al síndrome nefrítico agudo, salvo en que el inicio de la hematuria coincide con la enfermedad febril y se asocia a dolor lumbar. Es típica la proteinuria leve (<1 g/d), pero un £20% de los pacientes con nefropatía por IgA pueden desarrollar un SN. Siempre se produce una hematuria con presencia de hematíes dismórficos, pero los cilindros hemáticos son infrecuentes, al menos inicialmente. Aumenta la excreción urinaria de repligeno e interleucina 6. Las concentraciones séricas de creatinina y complemento suelen ser normales, pero la de IgA suele estar elevada. Puede producirse un aumento de los complejos fibronectina-IgA en suero, pero se duda de la importancia de este hallazgo. La hipertensión en el momento del diagnóstico es poco frecuente.
Diagnóstico
La hematuria macroscópica es la característica más habitual de la nefropatía por IgA y del síndrome de Alport, mientras que una hematuria microscópica persistente es más frecuente en la nefropatía por membrana basal delgada. La GN proliferativa mesangial se asocia con un patrón de lesión glomerular caracterizado por distintos grados de hipercelularidad mesangial o expansión de la matriz mesangial; con frecuencia se detectan depósitos de complejos inmunes a nivel mesangial. Muchas otras alteraciones heredofamiliares, inmunológicas e infecciosas (v. tabla 224-6) se ajustan a esta descripción morfológica.
Se puede distinguir la nefropatía por IgA de otras causas de hematuria renal primarias mediante estudios de inmunofluorescencia de la biopsia renal, que demuestran depósitos granulares de IgA y C3 en el mesangio expandido con focos de lesiones proliferativas o necrotizantes segmentarias. Sin embargo, también se pueden producir depósitos mesangiales de IgA en otras enfermedades (púrpura de Henoch-Schönlein, cirrosis hepática alcohólica).
Pronóstico y tratamiento
La nefropatía por IgA suele evolucionar lentamente; se desarrolla hipertensión e insuficiencia renal en 10 años en el 15 al 20% de los casos y en un 25% de los pacientes se produce una nefropatía terminal después de 20 años. Cuando se diagnostica una nefropatía por IgA en la infancia, el pronóstico suele ser bueno. Sin embargo, una hematuria persistente siempre se acompaña de hipertensión, proteinuria e insuficiencia renal. El debut en edades más avanzadas, la hipertensión, la proteinuria persistente grave, la ausencia de hematuria recidivante macroscópica, la elevación de los niveles de creatinina sérica y una esclerosis glomerular avanzada o la formación de semilunas y enfermedad tubulointersticial se consideran indicadores de mal pronóstico.
El tratamiento con IECA se comienza de manera precoz para la hipertensión y puede ser útil en los pacientes normotensos con proteinuria >1g/d. Si continúa el deterioro de la función renal, puede intentarse el tratamiento con aceite de pescado. Los glucocorticoides se reservan para la enfermedad de cambios mínimos demostrada por biopsia. Se requieren altas dosis iniciales (de 2 a 3 meses en adultos, 1 mes en niños), que luego se retiran de manera progresiva; únicamente el tratamiento de larga duración (2 años) es útil, y siempre deberán evaluarse los beneficios frente a su toxicidad. La globulina inmune i.v. (IG i.v.) puede ser útil en el tratamiento del trastorno funcional agudo (>2 ml/min/mes).
La GNRP que es tratada con pulsos i.v. de glucocorticoides seguidos de prednisona oral, ciclofosfamida oral o i.v., y/o plasmaféresis, evoluciona hacia una reducción de la concentración de la creatinina plasmática y de la proteinuria, pero no se da una reducción de las lesiones evidenciadas en la biopsia. En la mitad de los pacientes, la enfermedad progresa tras el cese del tratamiento.
SÍNDROME NEFRÍTICO-PROTEINÚRICO CRÓNICO
(Glomerulonefritis crónica, enfermedad glomerular lentamente progresiva)
Síndrome causado por diversas enfermedades de distintas etiologías, que se caracterizan morfológicamente por una esclerosis difusa de los glomérulos y clínicamente por proteinuria, cilindruria, hematuria y, en general, hipertensión, con una pérdida insidiosa de la función renal a lo largo de años.
Se ignora la incidencia en la población general, pero en las series de autopsias es del 0,5 al 1%.
Etiología
La etiología es diversa. Se reconoce una glomerulopatía primaria en un 50% de los pacientes con nefropatía terminal sometidos a nefrectomía bilateral y en un 50% de estos casos las alteraciones corresponden a una glomerulosclerosis focal y segmentaria en un 28%, a una GN inespecífica en un 28%, en un 25% a una GNRP, en un 15% una enfermedad con semilunas extensa y en 4% a unaglomerulonefritis membranosa grave (GNM).
La evidencia histológica de que las inmunoglobulinas y el complemento se distribuyen de forma variable en los glomérulos sugieren de forma indirecta una etiología inmune. Se han buscado posibles etiologías infecciosas, tóxicas o metabólicas, pero no se han encontrado. La coagulación intrarrenal ha sido implicada, ya que se han encontrado fibrinopéptidos que se derivan de la activación del sistema de la coagulación en la orina, la sangre y, en ocasiones, en el parénquima renal. Sin embargo, no se sabe si su presencia es causal o secundaria a las lesiones.
Morfología
En muchos glomérulos se observa un incremento de la matriz extracelular que engloba la matriz mesangial, la MBG y las asas capilares colapsadas, sin una hipercelularidad significativa. Se suelen producir sinequias glomerulares organizadas (adherencias epiteliales en el espacio de Bowman), que pueden llegar a afectar al 50% de la arquitectura glomerular. Los depósitos de inmunoglobulinas detectados por inmunofluorescencia son inconstantes y pueden faltar.
En función del estadio de la enfermedad se puede afectar el intersticio, aunque con frecuencia muestra una afectación precoz con infiltración y fibrosis extensas. También se produce atrofia tubular. Las lesiones vasculares son inespecíficas, recuerdan a los cambios nefroscleróticos y pueden producirse por hipertensión. Se debe sospechar una lesión renal progresiva e irreversible cuando se produzca una esclerosis glomerular grave y difusa, con sinequias en varios glomérulos, una enfermedad intersticial desproporcionada para el grado de lesión glomerular y un marcado aumento del material extracelular.
Signos y síntomas
El síndrome se mantiene asintomático durante años, por lo que no se detecta en la mayor parte de los casos. Los pacientes pueden comenzar con síntomas de uremia relacionados con una nefropatía terminal (náuseas, vómitos, disnea, prurito, fatiga). Durante toda la evolución del síndrome, se pueden producir recaídas de la hematuria macroscópica (infrecuente) y la proteinuria, lo que posiblemente representa una nefropatía IgA progresiva u otros tipos de hematuria renal idiopática, brotes de una enfermedad lentamente progresiva o episodios no relacionados de un síndrome nefrítico agudo. Se puede observar edema en zonas declives, generalmente asociado con una insuficiencia renal moderada por SN. Es frecuente la hipertensión de gravedad variable, que se suele acompañar de insuficiencia renal, aunque a veces antecede a la azoemia significativa.
Hallazgos de laboratorio
La proteinuria, que es un hallazgo constante, es generalmente de rango no nefrótico. Se suelen observar hematíes dismórficos y cilindros de hematíes, aunque pueden faltar en la enfermedad establecida. Habitualmente se detectan cilindros hialinos y de células tubulares fina o groseramente granulosos en cantidad moderada en el sedimento urinario, en función de la gravedad de la lesión. Sólo se observan cilindros anchos y céreos cuando existe una cicatrización intersticial significativa con atrofia tubular. Cuando se destruye al menos el 50% de la masa renal funcionante, aumentan el BUN y la creatinina séricos; al progresar la enfermedad aparecen anemia, acidosis metabólica, hiperfosfatemia y otras alteraciones bioquímicas que sugieren una insuficiencia renal crónica.
Diagnóstico y tratamiento
Esta enfermedad se puede descubrir durante una exploración médica rutinaria cuando el paciente se encuentra asintomático y la función renal es normal, salvo por la existencia de proteinuria y (posiblemente) hematuria. Antes de que se produzcan alteraciones significativas en la función renal, la biopsia puede contribuir a la distinción entre la hematuria recidivante idiopática, la enfermedad no glomerular (tubulointersticial) y el síndrome nefrítico-proteinúrico crónico. La glomerulosclerosis focal y segmentaria, la GNM, la GNRP y la nefropatía por IgA son los procesos que con más frecuencia se confunden con este síndrome. La biopsia renal no suele estar indicada cuando los riñones aparecen retraídos y con aspecto cicatrizal, ya que el estudio histológico suele aportar poca información etiológica en estos casos.
No se ha demostrado que ningún tratamiento evite la progresión. Un tratamiento antihipertensivo adecuado y una restricción correcta de Na pueden resultar útiles. La restricción en dieta de fosfatos y proteínas y los inhibidores de la ECA pueden retrasar el deterioro en otras patologías renales y puede ser útil en la mayor parte de las glomerulopatías crónicas. El tratamiento de la uremia se comenta en Insuficiencia renal crónica, capítulo 222.
SÍNDROME NEFRÓTICO
Complejo predecible que se produce por un aumento prolongado y grave en la permeabilidad glomerular para las proteínas.
La característica principal es una proteinuria grave (>2 g/m2/d o un cociente proteína/creatinina urinaria medido al azar >2), pero también se suele producir hipoalbuminemia (<3 g/dl), edema generalizado, lipiduria y lipidemia.
Etiología y clasificación
El síndrome nefrótico (SN) se puede producir a cualquier edad, pero su prevalencia es más elevada en los niños que en los adultos. En los niños afecta con más frecuencia a los de 1,5 a 4 años, con predilección por los varones, mientras que en adultos la distribución por sexos es similar. Se produce proteinuria por las alteraciones funcionales de dos mecanismos: la barrera selectiva por tamaños permite que pasen moléculas proteicas grandes y la barrera selectiva por carga no consigue retener las proteínas de bajo peso molecular. En la tabla 224-7 se enumeran las enfermedades que producen SN.

Entre las causas primarias destacan la enfermedad de cambios mínimos, la glomerulosclerosis focal y segmentaria (GEFS), la glomerulonefritis membranosa (GM), la glomerulonefritis membranoproliferativa (GNMP) y la glomerulonefritis proliferativa mesangial. El SN también se puede deber a causas secundarias.
Signos y síntomas
Un signo precoz de SN es una orina espumosa por las proteínas. Otras características incluyen la anorexia, el malestar, los párpados edematosos, el brillo retiniano, el dolor abdominal y la pérdida de masa muscular. Se puede producir anasarca con derrame pleural y ascitis.
Puede presentarse un edema focal que cursa con dificultad respiratoria (derrame pleural o edema de laringe), dolor torácico subesternal (edema pericárdico), tumefacción escrotal, hinchazón de las rodillas (hidrartrosis) y aumento de perímetro abdominal (ascitis). En los niños el dolor abdominal se puede relacionar con el edema mesentérico. Con frecuencia el edema es móvil (se detecta en los párpados por la mañana y en los tobillos tras deambular). El edema puede dificultar la valoración de la pérdida de masa muscular. Se pueden producir líneas blanquecinas paralelas en los lechos ungueales por el edema subungueal.
Los adultos pueden tener hipotensión, normotensión o hipertensión, en función de los niveles de angiotensina II. Se puede producir oliguria o insuficiencia renal aguda por la hipovolemia y la reducción de la perfusión renal. En niños se puede desarrollar hipotensión ortostática e incluso shock.
Complicaciones
El principal problema viene determinado por las consecuencias bioquímicas de la proteinuria grave. Un SN prolongado puede producir deficiencias nutricionales graves, incluida malnutrición proteica que recuerda al kwashiorkor, pelos y uñas frágiles, alopecia, retraso del crecimiento, desmineralización ósea, glucosuria, hiperaminoaciduria de diversos tipos, depleción de K+, miopatías, reducción del Ca total, tetania e hipometabolismo. Se puede producir una peritonitis espontánea y existe elevada prevalencia de infecciones oportunistas. Se considera que esta gran incidencia de infecciones guarda relación con la pérdida urinaria de inmunoglobulinas. Los trastornos de la coagulación con reducción de la actividad fibrinolítica e hipovolemia episódica suponen un riesgo de trombosis (sobre todo de la vena renal). Se suele producir hipertensión con las consiguientes complicaciones cerebrales y cardíacas en los pacientes con diabetes o enfermedades del colágeno.
Hallazgos de laboratorio
Orina. El análisis de orina muestra en fases iniciales una proteinuria intensa (>2 g/m2/d o un cociente proteína/creatinina urinaria determinado al azar >2). El sedimento urinario suele contener cilindros hialinos, granulosos, grasos, céreos o de células epiteliales. La lipiduria se determina realizando una tinción de Sudán de los cilindros que contienen gránulos de lípidos, identificando macrófagos o células tubulares renales que contengan gotas de grasa (cuerpos grasos ovales) y demostrando la presencia de cristales anisotropos (cuerpos grasos birrefringentes) con microscopio de polarización. También se pueden producir hematuria microscópica y cilindros hemáticos en función de la causa de la glomerulopatía. Los leucocitos son prominentes en caso de enfermedades exudativas y LES. Se pueden reconocer fibras de amiloide con microscopia electrónica en la nefropatía de la amiloidosis.
El K+ urinario suele estar elevado en la fase de acumulación del edema nefrótico; la concentración de Na urinaria suele ser inferior a 1 m mol/l (cociente Na+/K+ > 1). La secreción de aldosterona está elevada en esta fase, pero puede ser normal en otros momentos a pesar del edema mantenido. Las concentraciones de nitrógeno ureico y creatinina en suero varían en función del grado de alteración de la función renal.
Sangre. Se detecta hipoalbuminemia mediante determinación química o electroforesis cuantitativa. La albúmina suele ser <2,5 g/dl y en niños a veces es <1 g/dl. En la orina se pierden determinadas proteínas transportadoras y suelen disminuir los niveles de a-globulinas y g-globulinas, de otras inmunoglobulinas, de las hormonas tiroideas y de la corteza adrenal, de ceruloplasmina, de transferrina, de ASO y de complemento.
La lipemia se demuestra por el incremento en los niveles de colesterol y triglicéridos. Unos niveles de triglicéridos >10 veces los normales se relacionan con una hipoalbuminemia grave por aumento de la producción de lípidos y menor eliminación de los mismos.
Son frecuentes las coagulopatías, quizá por la pérdida urinaria de factor IX y XII y factores trombolíticos (urocinasa y antitrombina III) y el aumento de los niveles séricos de factor VIII, fibrinógeno y plaquetas.
Diagnóstico
El diagnóstico se sugiere ante las características clínicas y los hallazgos de laboratorio y se confirma con una biopsia renal. Para el diagnóstico hace falta una proteinuria grave, aunque la distinción entre la proteinuria nefrótica y no nefrótica es arbitraria. En cualquier caso, las enfermedades que afectan de manera preferente a la vasculatura extraglomerular, los túbulos o el intersticio no producen habitualmente una proteinuria muy intensa.
La insuficiencia renal no suele ser un síntoma de presentación en el SN, pero se puede producir tras una enfermedad prolongada. Sin embargo, los pacientes con SN debido a causas secundarias tienen generalmente una insuficiencia renal desde el principio o en fases tempranas. La presencia de una intensa proteinuria en un paciente nefrítico suele indicar que la enfermedad está avanzada y se considera un signo ominoso.
Se debe descartar en los pacientes la presencia de enfermedades sistémicas frecuentes (diabetes mellitus, amiloidosis, mieloma múltiple, LES). Si los pacientes refieren pérdida de peso o son ancianos, se deben descartar los procesos malignos y un posible efecto farmacológico. Otras causas de proteinuria en rango no nefrótico se recogen en el capítulo 214.
Pronóstico
El pronóstico depende de la causa. Se puede producir una remisión completa si el SN se debe a una enfermedad susceptible de tratamiento, algo que se produce en un 50% de los casos infantiles y con menos frecuencia en adultos. El pronóstico suele ser favorable en los casos que responden a esteroides (v. más adelante en Tratamiento). Algunas enfermedades causantes de SN remiten de forma espontánea, incluso después de 5 años.
En todos los casos el pronóstico empeora por las infecciones, la hipertensión, la azoemia significativa, la hematuria o las trombosis de las venas cerebrales, pulmonares, periféricas o renales. La incidencia de recidivas es alta en los pacientes trasplantados por GEFS, LES, nefropatía por IgA y, sobre todo, por GNRP tipo II, siendo menos frecuente en la de tipo I. También se producen recidivas en pacientes trasplantados por GNM y GN proliferativa mesangial.
Tratamiento
El tratamiento se orienta a corregir el proceso patogénico subyacente y depende de la morfología renal (v. tabla 224-3).
El tratamiento de soporte incluye una dieta con aproximadamente 1 g/kg/d de proteínas biológicas de alta calidad, pobre en grasa saturada y colesterol y rica en fibra. Sólo se necesitan suplementos de proteínas cuando el paciente esté malnutrido y sólo se debe restringir la ingesta de las mismas cuando exista una elevación de la creatinina sérica y la ingesta excesiva de proteínas agrave la proteinuria. Pueden ser necesarias estatinas hipolipemiantes (pravastatina, lovastatina, simvastatina, atorvastatina) para controlar la hipercolesterolemia; en estos pacientes se produce una incidencia elevada de rabdomiólisis, por lo que se debe controlar la creatinfosfocinasa. Los inhibidores de la ECA suelen controlar la proteinuria y la lipemia, pero pueden agravar la hiperpotasemia en los pacientes con una disfunción renal moderada o grave. Se debe iniciar un ejercicio físico gradual.
La ingesta de K+ debe ser de 1 mmol/kg/d. Si se produce hiponatremia, se debe limitar la ingesta de líquidos. Si se produce una diuresis breve y mejora el edema, se pueden relajar las limitaciones de la ingesta de Na+. Si existe ascitis, puede resultar útil realizar varias comidas pequeñas. Para controlar el edema sintomático se recomienda la restricción de Na+ (<100 mmol/d). Se pueden usar tiazidas o diuréticos del asa, aunque es posible que alteren la función renal y aumenten el riesgo de trombosis. Si la hipovolemia es grave y amenaza la vida, puede estar justificada la infusión de plasma o albúmina. Se debe tratar la hipertensión, en general con inhibidores de la ECA y diuréticos o en ocasiones con otros fármacos. La trombosis es frecuente y se debe descartar (sobre todo la trombosis venosa profunda y el embolismo pulmonar); los anticoagulantes profilácticos pueden resultar útiles cuando la albúmina sérica sea <2,5 g/dl o en presencia de tromboembolismo.
Las infecciones (sobre todo la bacteriuria, la endocarditis y la peritonitis) comprometen la vida y se deben detectar y tratar con rapidez. La eliminación de los antígenos infecciosos (endocarditis estafilocócica y por Streptococcus viridans, la nefritis sobre prótesis vascular, la malaria, la sífilis, la esquistosomiasis) puede curar el SN. Una cuidadosa desensibilización puede revertir el SN asociado con alergenos (hiedra venenosa, antígenos de insectos). La eliminación de las nefrotoxinas (oro, penicilamina, AINE) puede conseguir la remisión.
ENFERMEDAD CON CAMBIOS MÍNIMOS
(Nefrosis lipoide; enfermedad nula)
Enfermedad (especialmente infantil), que se caracteriza por edema de inicio brusco, proteinuria muy selectiva, con un sedimento urinario benigno, función renal normal y tensión normal.
Etiología y fisiopatología
La enfermedad con cambios mínimos (ECM) es la causa de SN más frecuente en niños de 4 a 8 años (90%), pero también afecta a adultos (20%). La prevalencia geográfica de las enfermedades predisponentes (esquistosomiasis en Egipto, malaria en Nigeria) se considera un factor importante en la prevalencia de este proceso.
La ECM se asocia con linfoma de células T, enfermedad de Hodgkin y nefritis tubulointersticial asociada a AINE. Se considera relacionada con una pérdida del equilibrio de las subpoblaciones de linfocitos T y con la alteración de la regulación de la inmunidad mediada por células, que produce una citocina con actividad sobre la permeabilidad. Esta actividad de la citocina disminuye cuando se produce la supresión de las células T en las infecciones virales (sarampión) y con fármacos esteroides o citotóxicos.
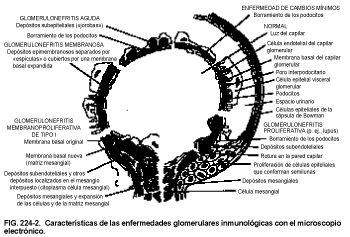
El cambio morfológico sólo es aparente con microscopía electrónica, que muestra edema con tumefacción difusa (borramiento) de los podocitos de las células epiteliales (v. fig. 224-2). Aunque este borramiento no resulta evidente cuando no existe proteinuria, se puede observar una proteinuria grave en presencia de podocitos normales. En un 5% de los casos se observa una hipercelularidad mesangial.
Signos, síntomas y diagnóstico
La ECM se caracteriza por un SN sin hipertensión ni azoemia; un 20% de los pacientes presentan hematuria microscópica. La azoemia se puede producir en los casos no idiopáticos y en pacientes >60 años. La proteinuria de la ECM es muy selectiva para la albúmina. En los niños, a no ser que existan características atípicas, el ensayo terapéutico con esteroides confirma el diagnóstico. Si existe resistencia a esteroides con defectos tubulares que producen glucosuria renal, aminoaciduria, acidosis tubular renal y pérdida de fosfatos, el diagnóstico puede ser GEFS más que de ECM. En los niños con resistencia a esteroides y en los adultos, el diagnóstico de este proceso se realiza mediante el estudio con microscopio electrónico de la biopsia renal.
Pronóstico y tratamiento
El pronóstico es bueno y se observan remisiones espontáneas en el 40% de los casos, siendo rara la progresión hacia la insuficiencia renal (5%).
Un 80 a 90% de los pacientes responden al tratamiento inicial con esteroides (60 mg/m2/d de prednisona durante 4 a 6 sem en niños y 1 a 1,5 mg/kg/d durante 6 a 8 sem en adultos, ambos v.o.), pero un 40 a 60% de los pacientes que responden recaen. Los pacientes que responden (cese de la proteinuria o diuresis si existe edema) deben seguir con la prednisona durante otras 2 sem y después iniciar un régimen de mantenimiento para reducir al mínimo la toxicidad (2 a 3 mg/kg en días alternos durante 4 a 6 sem en niños y 8 a 12 sem en adultos, disminuyendo las dosis a lo largo de 4 meses). Un tratamiento inicial más prolongado y un descenso lento de las dosis de prednisona reducen la incidencia de recaídas. Los pacientes que no responden a esteroides pueden tener una esclerosis focal asociada.
En los pacientes que no responden a esteroides (<5% de los niños y >10% de los adultos), en los que presentan recaídas frecuentes y en los enfermos dependientes de esteroides, se puede conseguir una remisión completa con un citotóxico oral (habitualmente ciclofosfamida, 2 a 3 mg/kg/d durante 12 sem o clorambucilo 0,15 mg/kg/d durante 8 sem). Sin embargo, estos fármacos pueden producir supresión gonadal (más grave en la adolescencia prepuberal) y la ciclofosfamida puede causar una cistitis hemorrágica y suprimir la médula ósea y la función linfocitaria. Se debe controlar la dosis realizando frecuentes hemogramas y descartar la cistitis hemorrágica mediante análisis de orina. Los adultos, sobre todo los ancianos o hipertensos, tienen mayor riesgo de complicaciones yatrogénicas por citotóxicos. Otra alternativa es la ciclosporina oral, 5 mg/kg/d divididas en 2 dosis, ajustando las dosis hasta conseguir una concentración valle en sangre completa de 150 a 300 mg/l (125 a 250 nmol/l) determinada mediante radioinmunoensayo con anticuerpos monoclonales. Se produce una remisión completa en >80% de los casos y se suele mantener el tratamiento durante 1 o 2 años para evitar las recaídas.
En los casos resistentes se puede aplicar un tratamiento alternativo con inhibidores de la ECA, tioguanina o levamisol o azatioprina a largo plazo. Se deben valorar los riesgos del tratamiento frente a los beneficios de reducción de la proteinuria, posiblemente sin modificar la evolución de las restantes manifestaciones del SN.
GLOMERULOSCLEROSIS FOCAL Y SEGMENTARIA
Enfermedad caracterizada por una proteinuria de inicio insidioso, con hematuria leve, hipertensión y azoemia, que se produce habitualmente en adolescentes, pero se puede observar también en adultos jóvenes o de mediana edad.
Etiología e histología
La glomerulosclerosis focal y segmentaria (GEFS) es la tercera causa más frecuente de SN en adultos (20 a 30%), siendo especialmente frecuente en varones de raza negra. La GEFS es idiopática, pero se relaciona con el uso de drogas i.v. y se produce en un 20% de los pacientes VIH positivos no homosexuales.
Las evidencias sugieren que un factor circulante no inmunoglobulínico determina el aumento de la permeabilidad glomerular a las proteínas, alteración que puede inducir la esclerosis asociada. La GEFS se inicia en los glomérulos yuxtamedulares (que pueden faltar en la muestra de biopsia). Se produce una hialinización segmentaria con depósitos de IgM y C3 con un patrón nodular o granular grueso y una pérdida difusa de los podocitos. Se puede producir una esclerosis global, que provoca atrofia glomerular.
Signos, síntomas y diagnóstico
Los pacientes con GEFS suelen debutar con hematuria, hipertensión y disfunción renal con SN, aunque en ocasiones la única clínica es una proteinuria asintomática en rango no nefrótico. La proteinuria es típicamente no selectiva. Los niveles de IgG suelen estar bajos. El diagnóstico se confirma mediante biopsia renal.
Pronóstico y tratamiento
El pronóstico es malo porque el tratamiento no es muy eficaz y las remisiones espontáneas son infrecuentes. Más del 50% de los pacientes desarrollan una insuficiencia renal en 10 años y en un 20% se produce una nefropatía terminal en 2 años. La enfermedad evoluciona con más rapidez en los adultos que en los niños. Cuando se produce pérdida de la luz capilar (variante con colapso), el pronóstico es malo. La gestación puede agravar la GEFS.
La GEFS recidiva en el 20 al 30% de los pacientes trasplantados, reapareciendo a veces la proteinuria en horas. Un 30 a 50% de los pacientes con GEFS recidivante pierden el injerto, riesgo que es más elevado en los niños pequeños, en los pacientes que desarrollan una insuficiencia renal en <3 años desde el inicio de la enfermedad y en los que tienen proliferación mesangial.
Los adictos a la heroína con SN por GEFS pueden tener una remisión completa del SN si dejan de consumirla en fases precoces de la enfermedad.
La GEFS puede remitir espontáneamente a veces o responder a esteroides (p. ej., 40 a 80 mg/d de prednisona durante 8 a 12 sem, seguidos de prednisona 30 a 40 mg/d en días alternos durante 8 a 12 sem). Si sólo se observa una ligera mejoría o se produce una recaída, se puede conseguir la remisión añadiendo ciclofosfamida (2 a 3 mg/kg/d durante 12 sem) o ciclofosfamida (5 mg/kg/d en adultos o 6 mg/kg/d en niños, durante 16 sem). En la enfermedad avanzada se prefiere el tratamiento prolongado con inhibidores de la ECA para reducir la proteinuria y retrasar la progresión de la enfermedad. Una alternativa la constituye la plasmaféresis con inmunosupresión con tacrólimo. El tratamiento enérgico puede reducir la insuficiencia renal progresiva pero aumenta el riesgo de complicaciones serias. Todavía no se ha establecido la utilidad de los anticoagulantes y antitrombóticos.
Nefropatía asociada al VIH
Esta variante de GEFS se suele observar en los pacientes VIH positivos de raza negra que consumen heroína, y es poco frecuente en la raza blanca. Con microscopio de luz se suele observar colapso capilar con túbulos dilatados, sin fibrosis intersticial ni atrofia tubular. En las células endoteliales se reconocen inclusiones reticulares tubulares parecidas a las que se observan en el LES.
La nefropatía asociada al VIH se puede producir en presencia de síntomas de SIDA o del complejo relacionado con el SIDA. Puede comenzar con azoemia leve o proteinuria grave; los riñones están aumentando de tamaño y son muy ecogénicos en la ecografía. Se produce una progresión rápida hacia la insuficiencia renal terminal en 3 a 4 meses. La normalidad de la tensión arterial y el aumento de tamaño renal mantenido distinguen esta variante.
El pronóstico es malo y depende de la enfermedad subyacente, siendo el tratamiento meramente paliativo y de sostén.
GLOMERULONEFRITIS MEMBRANOSA
Enfermedad (principalmente de adultos) caracterizada por un edema de inicio insidioso, con proteinuria intensa, con un sedimento urinario benigno, una función renal normal y una TA normal o alta.
Etiología e histología
La glomerulonefritis membranosa (GM) suele afectar a adultos y se considera la causa más frecuente de SN en los pacientes >40 años. Suele ser idiopática, pero se puede relacionar con fármacos (oro, penicilamina), infecciones (hepatitis B), enfermedades autoinmunes (LES) o procesos malignos (enfermedad de Hodgkin o linfomas no hodgkinianos, leucemia linfática crónica, carcinomas sólidos del pulmón, mama, colon, estómago o riñón y melanonas). Aunque es poco frecuente, la GNM en niños se suele asociar con el virus de la hepatitis B y el LES.
Los complejos inmunes se visualizan como depósitos densos con microscopía electrónica (v. fig. 224-2). Se producen depósitos subepiteliales densos en fases iniciales de la enfermedad, con espículas (spikes) de lámina densa entre los depósitos. Posteriormente los depósitos se localizan dentro de la membrana basal glomerular (MBG), con marcado engrosamiento de la misma. Se producen depósitos de IgG difusos y granulares a lo largo de la MBG, pero no hay fenómenos de proliferación celular, exudación ni necrosis.
Signos, síntomas y diagnóstico
El inicio del SN puede ser insidioso. Es característico que comience con edema y proteinuria en rango nefrótico y, en ocasiones, se presenta hematuria microscópica e hipertensión. Sólo un 20% de los pacientes tienen proteinuria de rango no nefrótico. Los niveles de C3 y C4 son normales. El IFG es normal o bajo. Las proteínas urinarias muestran una selectividad variable. En el momento inicial se pueden apreciar síntomas de las enfermedades asociadas (enfermedades por complejos inmunes o enfermedades del colágeno, infecciones crónicas, neoplasias). Se debe buscar una neoplasia oculta (en el 2% de los casos), sobre todo si el paciente refiere pérdida de peso, tiene una anemia no explicable o sangre oculta en heces o es anciano. También se debe descartar una GNM secundaria a fármacos. Entre las complicaciones destacan la trombosis de la vena renal, el embolismo, la nefritis intersticial y la glomerulonefritis rápidamente progresiva (GNRP). El diagnóstico se confirma mediante biopsia renal.
Pronóstico y tratamiento
El curso es relativamente benigno en la mayor parte de los casos; un 25% remiten de forma espontánea, un 25% desarrollan una proteinuria en rango no nefrótico mantenida, un 25% desarrollan un SN persistente y el 25% restante evolucionan hacia una nefropatía terminal. Las mujeres, los niños y adultos jóvenes con proteinuria en rango no nefrótico y los que conservan una función renal normal a los 3 años del diagnóstico muestran poca tendencia a la progresión.
Los pacientes asintomáticos con proteinuria en rango no nefrótico no deben recibir tratamiento, dado su buen pronóstico a largo plazo. Sin embargo, se debe controlar la función renal periódicamente para descartar la progresión del proceso. Los pacientes con proteinuria en rango nefrótico asintomáticos o que presenta edema controlable con diuréticos deben ser vigilados, ya que al menos un 50% de los mismos muestra una remisión completa o parcial en 3 o 4 años.
Se debe considerar la administración de fármacos inmunosupresores sólo en los pacientes con SN sintomáticos y en los que tengan más riesgo de progresión: varones >50 años con proteinuria de al menos 10 g/d, los pacientes con un incremento de la b-microglobinuria y los que tengan un incremento de la creatinina sérica inicialmente.
Parece que resulta más eficaz administrar los fármacos citotóxicos diariamente por vía oral que en pulsos intravenosos (80% de remisiones parciales o completas). En general se administran ciclofosfamida (1,5 mg/kg/d) o clorambucilo (0,2 mg/kg/d) durante 6 a 12 meses junto con prednisona (60 mg/d o 120 mg en días alternos). En los pacientes que no toleran los fármacos citotóxicos, se pueden conseguir beneficios con ciclosporina, 4 a 6 mg/kg/d durante 4 meses. Entre los tratamientos propuestos, cuya eficacia a largo plazo no se ha demostrado, destacan los inhibidores de la ECA, la inmunoglobulina i.v. y los AINE.
GLOMERULONEFRITIS MEMBRANOPROLIFERATIVA
Enfermedad poco frecuente (sobre todo infantil) en la que se produce SN con hematuria microscópica y, con menos frecuencia, síndrome nefrítico agudo.
Etiología e histología
La glomerulonefritis membranoproliferativa (GNMP) parece un grupo heterogéneo de enfermedades de causa inmunológica. Algunos datos apoyan la predisposición genética, ya que los familiares o hermanos de los pacientes con anemia drepanocítica, lipodistrofia parcial, deficiencia congénita de complemento o síndrome de Down presentan un riesgo mayor.
Aunque se trata de una enfermedad renal primaria típica, la GNMP se puede producir en pacientes con enfermedades sistémicas (LES, crioglobulinemia mixta, hepatitis crónica activa), con neoplasias (leucemia linfática crónica, linfomas, nefroblastomas) o trastornos infecciosos (endocarditis bacteriana, abscesos viscerales, VIH, malaria, esquistosomiasis). Existen dos variantes de GNMP en función de las alteraciones ultraestructurales del glomérulo: tipo I (GN mesangiocapilar), que representa un 80 a un 85% de los casos, y tipo II (enfermedad por depósitos densos), que representa el 15 al 20% restante.
Este proceso afecta por igual a varones y mujeres, siendo más frecuente en los pacientes de raza blanca que en las restantes razas. Durante las 2 últimas décadas, se ha producido un descenso significativo en la incidencia de la tipo I por la mejora en las condiciones de vida, los mejores hábitos higiénicos y ambientales y la reducción de la incidencia y la gravedad de las infecciones. El tipo II se produce sobre todo en pacientes jóvenes y su incidencia no ha cambiado, siendo su patogenia desconocida.
En la GNMP tipo I se producen depósitos sub-endoteliales electrodensos de IgG, IgM y C3 predominantemente, y la matriz mesangial se sitúa entre la MBG y las células endoteliales. La duplicación de la MBG determina la clásica imagen en «raíl» o doble contorno (v. fig. 224-2). En la GNMP tipo II, los depósitos electrodensos que contienen C3 reemplazan parcialmente la lámina densa con un aspecto festoneado característico, originando un engrosamiento de la MBG. La glomerulosclerosis que produce la insuficiencia renal en ambos tipos de GNMP parece determinada por el depósito crónico de complejos inmunes en el glomérulo, tanto en la pared capilar como en el mesangio. La activación de la vía clásica del complemento se produce en la GNMP tipo I, mientras que en la tipo II se activa por vía alternativa.
Síntomas, signos y diagnóstico
La GNMP se asocia con SN en un 60 a un 80% de los casos y se suele acompañar de hematuria microscópica. Hasta en el 30% de los pacientes se puede producir una proteinuria de rango no nefrótico y del 10 al 20% comienzan con nefritis aguda. En algunas ocasiones se producen hematuria macroscópica, azoemia e hipertensión. Los pacientes con GNMP pueden ser asintomáticos, con hematuria y proteinuria exclusivamente. Son típicas las fluctuaciones espontáneas de la gravedad clínica.
La hipocomplementemia es producida por muchos factores y se considera sólo un marcador. Se presenta frecuentemente una anemia normocítica normocroma inesperada en el estadio de la insuficiencia renal presente. En la mayoría de los pacientes se produce un consumo de plaquetas con activación del potente factor de crecimiento derivado de las plaquetas. Hay que realizar pruebas serológicas para LES, hepatitis B y C y crioglobulinemia mixta para descartar enfermedades sistémicas que son causa frecuente de GNMP.
El diagnóstico depende de los hallazgos de la biopsia renal. En la GNMP tipo I la insuficiencia renal se suele asociar con una elevación de la creatinina sérica y con la presencia de lesiones tubulointersticiales en el momento de la biopsia, seguidas de una hipertensión persistente con proteinuria grave. En general no se puede diferenciar la presentación de la tipo I y la tipo II, aunque parece que el síndrome nefrítico agudo es más frecuente en la segunda.
Pronóstico y tratamiento
La GNMP de tipo I suele progresar con lentitud y la de tipo II más deprisa. En general el pronóstico a largo plazo es malo y se produce una nefropatía terminal en el 50% de los pacientes a los 3 a 5 años y en el 75% a los 10 años; a los 5 años sólo un 25% de los pacientes conservan una función renal normal. Se producen remisiones espontáneas en <5% de los casos. La GNMP tipo I recidiva en un 30% de los trasplantes renales y la tipo II en el 90%.
Probablemente no está indicado un tratamiento específico en los pacientes con proteinuria en rango no nefrótico, ya que el pronóstico a largo plazo es relativamente benigno. En los niños nefróticos el tratamiento con 2,5 mg/kg de prednisona a días alternos (máximo 80 mg) durante 1 año, seguido de un descenso de dosis hasta 20 mg, dosis que se mantiene durante 3 a 10 años, puede estabilizar la función renal. Sin embargo, el tratamiento con esteroides puede retrasar notablemente el crecimiento y producir hipertensión y en los adultos no consigue beneficios significativos. En los adultos la administración de dipiridamol (225 mg/d) con aspirina (975 mg/d) durante 1 año estabiliza la función renal 3 a 5 años, aunque los resultados a largo plazo no son evidentes. Se puede necesitar un tratamiento prolongado.
Entre las alternativas de tratamiento destacan el a-interferón con esteroides para la enfermedad producida por virus de la hepatitis C y la plasmaféresis para la crioglobulinemia grave o la GNRP. Los inhibidores de la ECA reducen la proteinuria y permiten controlar la hipertensión.
GLOMERULONEFRITIS PROLIFERATIVA MESANGIAL
Enfermedad que cursa con grados variables de proteinuria y con frecuencia con hematuria en el momento de presentación, que se inicia por el depósito o formación de complejos inmunes en el mesangio.
Etiología e histología
Aunque se ignora la incidencia, esta enfermedad aparece en el 5 al 10% de las biopsias renales realizadas por proteinuria, hematuria o azoemia. La GN proliferativa mesangial se caracteriza por un aspecto morfológico normal con microscopio de luz, mientras que la inmunofluorescencia y el microscopio electrónico demuestran aumento de celularidad y matriz mesangial que contiene IgM o IgA y C3.
Signos, síntomas y diagnóstico
Se producen grados variables de proteinuria, asociada con frecuencia con hematuria. Se observa SN en >75% de los casos, hematuria microscópica en el 20% e hipertensión leve en el 35%. Del 25 al 50% de los pacientes presentan azoemia moderada en el momento del diagnóstico. Existen complejos inmunes circulantes en el 50 al 70% de los casos. Pocos datos clínicos o de laboratorio permiten distinguir este trastorno de otras causas de proteinuria asociadas o no con hematuria. Se deben descartar la nefritis mesangial lúpica, la nefropatía por IgA, la púrpura de Henoch-Schönlein y la GN postinfecciosa en fase de resolución. El diagnóstico se realiza analizando la biopsia renal.
Pronóstico y tratamiento
Los pacientes que presentan una proteinuria grave tienen peor pronóstico porque suelen evolucionar hacia la insuficiencia renal y, en algunos casos, hacia la nefropatía terminal. Otros indicadores de mal pronóstico en el momento del diagnóstico incluyen la azoemia, la hematuria microscópica, la glomerulosclerosis focal y segmentaria y un aumento notable de la matriz mesangial con hipercelularidad.
Los adultos con proteinuria grave pueden responder a dosis altas de prednisona oral (60 mg diarios o 120 mg en días alternos). Aunque se pueden producir remisiones en el 20 al 60% de los casos, no son infrecuentes las recaídas, las remisiones parciales ni la dependencia de glucocorticoides. Si se produce hematuria es más probable la falta de respuesta a esteroides. En los niños que no responden a esteroides es frecuente la remisión tardía del SN.
SÍNDROMES NEFRÓTICOS CONGÉNITOS
La esclerosis mesangial difusa es rara, pero en EE. UU. es más frecuente que el tipo finlandés. Se desconoce el patrón de herencia. Un paciente con proteinuria grave debe ser sometido a una nefrectomía bilateral y debe iniciarse una diálisis precoz para disminuir las deficiencias nutricionales y la dificultad para crecer. Esta enfermedad suele recidivar en los riñones trasplantados.
El SN de tipo finlandés es autosómico recesivo y rápidamente progresivo y suele obligar a dializar en 1 año. La mayoría de los pacientes fallecen al cabo de 1 año, pero algunos han sobrevivido con soporte nutricional hasta que desarrollan una insuficiencia renal y entonces se tratan con diálisis o trasplante.
ENFERMEDADES MULTISISTÉMICAS QUE CURSAN CON SÍNDROME NEFRÓTICO
Varias enfermedades cursan con proteinuria marcada (v. tabla 224-7). Se comentan datos sobre la nefropatía diabética y la nefritis lúpica dada su gran importancia clínica.
Nefropatía diabética
(V. también Signos y síntomas en Diabetes mellitus, cap. 13.)
Epidemiología y patogenia
La nefropatía diabética (ND) es la causa más frecuente de nefropatía terminal en EE. UU. Posiblemente se subestima la prevalencia de insuficiencia renal en pacientes con diabetes mellitus de tipo II, pero se calcula que es de un 20 a un 30% y es más frecuente en pacientes de raza negra, asiáticos e hispanos con diabetes mellitus de tipo II. Del 20 al 30% de los pacientes con nefropatía terminal padecen una diabetes de tipo I y un 70 a 80% de tipo II.
La patogenia es compleja, pero los mecanismos responsables pueden ser hormonales y metabólicos así como las alteraciones hemodinámicas renales con hiperfunción e hipertrofia. La hiperfiltración, una alteración funcional precoz, se considera un elemento predictivo relativo del riesgo de desarrollar una insuficiencia renal.
La hiperglucemia produce una glucosilación de las proteínas glomerulares, lo que puede originar la proliferación de las células mesangiales, la expansión de la matriz y la lesión endotelial. La MBG se engrosa de forma característica.
Las lesiones de glomerulosclerosis difusa o nodular intercapilar son características. Se produce una hialinosis marcada de las arteriolas aferentes o eferentes y aterosclerosis, pudiendo existir también atrofia tubular y fibrosis intersticial. Sólo la expansión de la matriz mesangial parece relacionarse con la progresión a una nefropatía terminal.
Diagnóstico
La ND progresa en 10 a 25 años. Inicialmente se produce una hiperfiltración sin microalbuminuria que evoluciona a un IFG del 20 al 50% del normal y una microalbuminuria >300 mg/24 h. El IFG se normaliza en las lesiones renales precoces y la hipertensión leve y evoluciona a una hipertensión franca con proteinuria >0,5 g/d. La proteinuria grave y una pérdida progresiva de la función renal anteceden al desarrollo de una nefropatía terminal. Otras alteraciones de las vías urinarias son la necrosis papilar, la vejiga neurógena y la hidronefrosis con obstrucción funcional, los abscesos perirrenales, la pielonefritis aguda, la bacteriuria y la cistitis. Se produce con frecuencia una acidosis tubular renal de tipo IV.
El diagnóstico se sospecha ante una proteinuria, la coexistencia de una retinopatía diabética, hipertensión y los antecedentes de diabetes mellitus de >10 años de evolución. Se deben descartar otras patologías renales si se produce una intensa proteinuria en un paciente diabético de corta evolución, con hematuria macroscópica, con cilindros de hematíes o con una pérdida rápida del IFG. La biopsia renal confirma el diagnóstico.
Pronóstico y tratamiento
El tratamiento precoz puede retrasar la evolución de la enfermedad. Aunque la hipertrofia renal con hiperfiltración y las alteraciones morfológicas son típicas, no permiten saber qué pacientes van a desarrollar una ND y necesitan un tratamiento rápido. Entre los factores que permiten predecir la evolución de la ND y la necesidad de tratamiento destacan la microalbuminuria y la hipertensión. Unos niveles de TA en el límite alto de la normalidad (<140/90 mm Hg) o ligeramente elevados (<150/95 mm Hg) pueden acelerar el daño renal.
Los antecedentes de enfermedad vascular grave (antecedentes de ictus), de IM o gangrena periférica permiten predecir una supervivencia más corta. El SN suele anteceder a la nefropatía terminal en 3 a 5 años.
Es necesario un tratamiento enérgico. La restricción proteica retrasa la evolución a nefropatía terminal, posiblemente porque modifica la hemodinámica intrarrenal y reduce el IFG. La restricción de Na y fósforo reduce la progresión a insuficiencia renal. Una dieta restrictiva típica contiene 0,6 g/kg de proteínas, 2 g de Na, 0,5 a 1 g de fósforo y 1 g de Ca. El control de la glucemia (Hb glucosilada <7,5%) reduce la microalbuminuria pero no retrasa la progresión de la enfermedad una vez establecida la ND.
Los inhibidores de la ECA (p. ej., captopril) reducen la progresión de la nefropatía por sus efectos renoprotectores y antiproteinúricos. También están indicados en los pacientes normotensos con enfermedad renal incipiente. Si el desarrollo de tos persistente o hiperpotasemia impide la utilización de estos fármacos, se pueden emplear ciertos bloqueantes de los canales de Ca (como diltiazem o verapamilo) por sus parecidos efectos antiproteinúricos y renoprotectores. Los bloqueantes de los canales de Ca del grupo de las dihidropiridinas (nifedipino, felodipino y amlodipino) se deben evitar en los diabéticos porque pueden empeorar la proteinuria y la función renal. Los inhibidores de la ECA y los bloqueantes de los canales de Ca no pertenecientes al grupo de las dihidropiridinas tienen un efecto antiporteinúrico y renoprotector más importantes cuando se usan combinados y su efecto antiproteinúrico aumenta cuando se acompaña de restricción de sal.
El péptido C y los análogos de la somatostatina pueden ser útiles para controlar las complicaciones de la diabetes.
Los pacientes con nefropatía terminal por diabetes son candidatos a un posible trasplante. La tasa de supervivencia a los 5 años de los diabéticos de tipo II sometidos a trasplante renal es casi del 60%, frente al 2% en los pacientes dializados. La supervivencia del injerto es >85% a los 2 años. Sin embargo, en estos estudios no se tuvo en cuenta a los pacientes con complicaciones diabéticas graves con un mayor riesgo de muerte. El trasplante combinado riñón-páncreas ha tenido éxito en los pacientes con complicaciones diabéticas graves y progresivas.
Glomerulonefritis asociada a lupus eritematoso sistémico
Incidencia y morfología
La GN lúpica se puede asociar con síndrome nefrítico (v. tabla 224-2), pero se relaciona con más frecuencia con un síndrome nefrótico. En un 50% de los pacientes con lupus se observan alteraciones en el análisis de orina y azoemia. La incidencia total posiblemente supere el 90% porque las biopsias renales realizadas en pacientes con sospecha de LES sin evidencia clínica de nefropatía suele demostrar GN proliferativa focal o difusa.
La mayor parte de los pacientes presentan una GN mediada por complejos inmunes. Esta enfermedad se puede clasificar morfológicamente como mesangial (10 a 20%), proliferativa focal (10 a 20%), proliferativa difusa (40 a 70%) y membranosa (10 a 20%). La histología y las características clínicas y pronósticas son distintas en cada categoría, aunque existe un notable solapamiento entre ellas.
Los complejos inmunes en la GN lúpica están constituidos por antígenos nucleares (sobre todo ADN), anticuerpos antinucleares IgG con gran afinidad para fijarse con el complemento y anticuerpos contra el ADN. Son característicos los depósitos subendoteliales, intramembranosos y subepiteliales. En todos los lugares donde se depositan complejos inmunes, existe inmunofluorescencia positiva para complemento, IgA, IgG e IgM en cantidades variables.
Dada la limitada capacidad renal de responder a las lesiones, los distintos tipos de GN lúpica se parecen a otras glomerulopatías. Por ejemplo, la GN lúpica proliferativa difusa y membranosa se parecen morfológicamente a la glomerulonefritis membranosa idiopática y la GN membranoproliferativa de tipo I, respectivamente. Los datos de inmunofluorescencia y microscopía electrónica que sugieren nefropatía lúpica son la presencia de estructuras tubulorreticulares en las células endoteliales y de depósitos inmunes a lo largo de las membranas basales tubulares y de los vasos sanguíneos de pequeño calibre.
Signos, síntomas y diagnóstico
La mayoría de las alteraciones renales se producen durante el primer año desde el diagnóstico de LES; está indicado descartar una nefropatía cuando se sospecha o se ha diagnosticado un LES. El signo más frecuente es la proteinuria, aunque también se observa hematuria microscópica y cilindros de hematíes. La creatinina sérica suele aumentar de forma progresiva durante los primeros años de enfermedad. El descenso de los niveles séricos de complemento o la elevación de los títulos de anticuerpos anti-ADN sugieren una mayor actividad inmunológica y una posible progresión renal. La biopsia renal es esencial para determinar el tipo de GN lúpica, el pronóstico y el tratamiento.
Pronóstico y tratamiento
Las pruebas de laboratorio aisladas son indicadores escasamente fiables de curso adverso, aunque el descenso de los niveles de complemento o el aumento de los títulos de anticuerpos anti-ADN obligan a controlar de forma estrecha los datos que evidencian la progresión de la nefropatía. Un sedimento nefrítico, una proteinuria grave o un aumento progresivo de la creatinina sérica se consideran signos ominosos e indican la necesidad de tratamiento. En la biopsia renal, la presencia de proliferación de alto grado, la necrosis fibrinoide, la presencia de semilunas celulares o la existencia de extensos depósitos inmunes subendoteliales se consideran signos de mal pronóstico e indicativos de la necesidad de tratamiento intensivo. El pronóstico y los criterios para tratar la GN membranosa asociada al LES están mal definidos y son controvertidos.
El tratamiento suele combinar fármacos citotóxicos y esteroides. Se suele administrar ciclofosfamida i.v. en bolo (una vez al mes durante 6 meses y después una vez al trimestre durante 6 meses), empezando con dosis 0,75 g/m2 y, suponiendo que el recuento leucocitario sea >3.000/ml, aumentándola hasta un máximo de 1 g/m2 en una solución de suero salino en 30 a 60 min. Se administran 60 a 80 mg/d de prednisona, reduciendo la dosis hasta 20 a 25 mg en días alternos en 6 a 12 meses. Se suelen administrar ciclofosfamida y prednisona hasta que se consigue mantener la remisión de la nefritis por lo menos 1 año. Las recaídas se suelen tratar con dosis mayores de prednisona.
Una alternativa posiblemente más segura que la ciclofosfamida en el tratamiento de mantenimiento es la azatioprina oral (2 mg/kg/d), que se asocia a un riesgo menor de neoplasias tardías y no produce ningún riesgo de disfunción gonadal. También se puede emplear la azatioprina como tratamiento inicial en los pacientes que no quieren ser tratados con ciclofosfamida o no la toleran.