261 / MALFORMACIONES CONGÉNITAS
Defectos de la anatomía presentes desde el nacimiento.
Las malformaciones congénitas pueden ser hereditarias o esporádicas, únicas o múltiples, evidentes u ocultas, macroscópicas o microscópicas. Son la causa de casi la mitad de todas las muertes de los recién nacidos (RN) a término. Entre el 3 y el 4% de todos los RN presentan una malformación importante al nacer; hasta el 7,5% de todos los niños manifiestan los síntomas de un defecto congénito hacia los 5 años de edad.
La incidencia depende del tipo de malformación, del área geográfica del niño, lo que probablemente se explica por los factores genéticos, medioambientales o ambos (la espina bífida muestra una frecuencia de 3 a 4/1.000 nacimientos en ciertas áreas de Irlanda pero sólo alcanza 1/1.000 en Estados Unidos) y las costumbres (la consanguinidad aumenta el riesgo de anomalías congénitas). Con la edad de la madre (y, en menor medida, también del padre) asciende el riesgo de defectos cromosómicos, especialmente del síndrome de Down (v. tabla 247-1).
En la etiología pueden intervenir factores genéticos, teratógenos o de ambos tipos. Factores distintos que actúen en el mismo período de la organogénesis pueden causar defectos idénticos. Los factores genéticos producen tanto malformaciones aisladas como síndromes y pueden operar bien mediante una herencia mendeliana simple,bien mediante una herencia multifactorial. Algunos síndromes, como el de Down, se deben aalteraciones cromosómicas (v. más adelante, Anomalías cromosómicas). Los factores teratógenos pueden ser toxinas medioambientales, las radiaciones, la dieta, los fármacos, las infecciones y los trastornos metabólicos.
Diagnóstico
La ecografía, la amniocentesis y la biopsia corial (v. cap. 247) facilitan el diagnóstico de algunas malformaciones. Entre los factores obstétricos que sugieren la presencia de una anomalía congénita se encuentran la presentación de nalgas, el polihidramnios, que puede deberse a que el feto tiene dificultades para tragar (p. ej., por un trastorno grave del SNC, como la anencefalia) o a un bloqueo del aparato GI (p. ej., por atresia esofágica) y el oligo-amnios, que puede ser secundario a una escasa producción de orina por una alteración GU.
Tratamiento
Cuando se diagnostica una malformación grave antes del nacimiento, los padres pueden decidir terminar ese embarazo. En teoría, es posible hacer un tratamiento prenatal de los trastornos obstructivos (uropatía e hidrocefalia), pero estos procedimientos son todavía experimentales.
Cuando la malformación se diagnostica en el momento del nacimiento o poco después, los padres deben ser informados de inmediato, aunque la explicación detallada se posponga hasta consultar a los especialistas. La familia debe recibir una valoración realista de la gravedad del proceso, su pronóstico y la atención médica de que puede disponerse y debe participar activamente en las decisiones al respecto (v. también cap. 257).
Si se sospecha un factor genético, los padres deberán recibir el oportuno consejo genético (v. cap. 247).
CARDIOPATÍAS CONGÉNITAS
Defectos anatómicos del corazón y de los grandes vasos debidos a alteraciones producidas en distintas fases del desarrollo fetal y presentes en el momento del nacimiento.
Epidemiología y etiología
La incidencia es de 1/120 nacidos vivos. El riesgo se calcula en 2 a 3% de todos los niños con un pariente de primer grado afectado (mayor cuando el pariente es uno de los progenitores).
A veces existe una causa identificable. Algunas anomalías cromosómicas (p. ej., las trisomías 13 o 18) producen graves malformaciones cardíacas, mientras que otras (p. ej., la trisomía 21, el síndrome de Turner [XO] y los trastornos genéticos (p. ej., el síndrome de Holt-Oram) pueden dar lugar a procesos menos graves. También pueden estar implicadas las enfermedades maternas (p. ej., diabetes mellitus, LED, rubéola) o la exposición a factores medioambientales (talidomida, isotretinoína, alcohol [síndrome de alcohol fetal]) o sus combinaciones.
Fisiología y fisiopatología
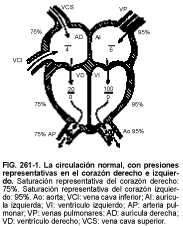
En el corazón normal, los cambios cardiovasculares que tienen lugar después del nacimiento (cierre del foramen oval y del conducto arterioso, disminución de la resistencia vascular pulmonar) separan las circulaciones sistémica y pulmonar (v. fig. 261-1 y Fisiología perinatal, cap. 256). Las presiones del corazón derecho son menores que las del izquierdo. Las consecuencias de las cardiopatías congénitas dependen de estas diferencias de presión.
Muchas cardiopatías congénitas no producen alteraciones hemodinámicas significativas; otras causan una sobrecarga anormal de volumen en alguno de los ventrículos, sobrecarga de presión intraventricular y una mezcla auricular de sangres oxigenada y no oxigenada, o un gasto cardíaco sistémico insuficiente.
Los defectos que suponen una obstrucción al flujo sanguíneo (p. ej., estenosis aórtica o pulmonar) producen soplos independientes de la caída de la resistencia vascular pulmonar. En general, estos soplos de eyección son ya audibles en el RN y poseen una calidad de incremento/decremento debida al aumento de la presión ventricular que se produce durante la sístole para superar la obstrucción. La hipertrofia ventricular, identificable mediante el ECG pero, habitualmente, no por radiografía, refleja el aumento consiguiente del trabajo ventricular.
Los cortocircuitos izquierda-derecha dependen de la disminución de la resistencia pulmonar, por lo que no suelen ponerse de manifiesto hasta que transcurre cierto tiempo después del nacimiento, variable entre varios días y unas pocas semanas, más breve en los de alta presión (es decir, en los ventrículos o en las grandes arterias) y mucho más largo para los de baja presión o auriculares. La dilatación ventricular, secundaria a los cortocircuitos izquierda-derecha, resulta evidente en las radiografías pero es menos visible en el ECG.
La insuficiencia cardíaca (v. más adelante) puede ser secundaria al aumento del flujo pulmonar (fácilmente identificable mediante la radiografía de tórax), que puede aumentar la presión venosa pulmonar.
Síntomas y signos
Los soplos y frémitos cardíacos debidos a la turbulencia del flujo sanguíneo en el corazón o en los grandes vasos son más marcados en las superficies próximas a su lugar de origen, lo que facilita su localización y diagnóstico. El incremento del flujo a través de la válvula pulmonar produce un soplo blando similar a los soplos de eyección aórtico o, sobre todo, pulmonar, aunque menos rudo. La sangre que regurgita a través de una válvula auriculoventricular o que atraviesa una comunicación interventricular produce un soplo pansistólico que a veces oculta los ruidos cardíacos normales a medida que su intensidad aumenta. El flujo en las grandes arterias es continuo, por lo que los soplos procedentes de las arterias aorta o pulmonar serán continuos y no estarán interrumpidos por los ruidos cardíacos. La calidad de los ruidos cardíacos refleja la función ventricular y las presiones de cierre arteriales. Cuando existen limitaciones a la apertura de la válvula, es fácil oír un chasquido de eyección que sigue al primer ruido cardíaco (S1).
Los signos de insuficiencia cardíaca pueden consistir en sufrimiento respiratorio con disnea y taquipnea, taquicardia y hepatomegalia. En el RN, el signo de presentación puede ser una cianosis. Las acropaquias y la policitemia se deben a una insaturación arterial de larga duración.
La insuficiencia de la perfusión sistémica se manifiesta por pulsos reducidos o impalpables, frialdad de las extremidades, escaso llenado capilar y, cuando este llenado es muy lento, signos de disfunción visceral (p. ej., disminución del gasto urinario, insuficiencia renal). El aumento del trabajo cardíaco puede dar lugar a hipertrofia de las cámaras del corazón.
Diagnóstico
El diagnóstico depende de la identificación correcta de la función cardíaca anormal (descrita en párrafos anteriores). La anamnesis, la exploración física, el ECG y las radiografías de tórax suelen ser suficientes para hacer un diagnóstico anatómico específico. La ecocardiografía, el cateterismo cardíaco, la angiocardiografía y otros estudios de laboratorio permiten confirmar el diagnóstico y establecer la gravedad del proceso.
La cianosis obliga a hacer una búsqueda meticulosa de las posibles causas no cardíacas. Muchos problemas respiratorios del período neonatal se manifiestan por cianosis cuando se asocian a obstrucciones del árbol bronquial, cuando existen grandes masas que ocupan el espacio habitualmente destinado al pulmón, o cuando una enfermedad alveolar impide el intercambio correcto de los gases. La hipotermia, la hipoglucemia, la hipocalcemia, la sepsis y la disfunción del SNC también suelen manifestarse por cianosis en el RN. La presencia de cianosis depende de la cantidad de Hb insaturada; por tanto, en ocasiones puede estar enmascarada por la anemia. El signo de Arlequín (eritema transitorio que afecta a un lado del cuerpo con palidez en el opuesto y una clara separación de ambos en la línea media), que remeda la cianosis, no es un fenómeno raro en los RN o lactantes pequeños.
COMUNICACIÓN INTERAURICULAR
Presencia de un orificio en el tabique interauricular.
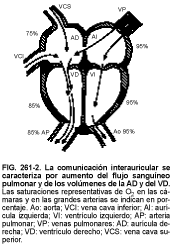
La comunicación interauricular constituye alrededor del 6 al 10% de todas las cardiopatías congénitas. Es más frecuente en la mujer (2:1) y se encuentra en 1:1.500 nacidos vivos. Se divide en dos tipos (v. fig. 261-2), ostium secundum y defectos del seno venoso.
Síntomas, signos y diagnóstico
El típico soplo de flujo de grado 2 a 3/6 es audible en el reborde esternal superior izquierdo, con desdoblamiento del segundo ruido cardíaco (S2) durante la respiración. Este soplo suele encontrarse >1 año de edad, cuando el flujo sanguíneo pulmonar ha aumentado ya considerablemente. Los grandes cortocircuitos izquierda-derecha (cociente flujo pulmonar: flujo sistémico ³2:1) pueden producir soplos diastólicos de tono bajo, de origen tricuspídeo. La radiografía muestra cardiomegalia, con dilatación de la aurícula derecha, del ventrículo derecho (VD) y de la arteria pulmonar, así como aumento del flujo sanguíneo en los pulmones. El ECG revela una moderada desviación del eje hacia la derecha, una sobrecarga también moderada de volumen en el VD, fuerzas ventriculares izquierdas normales y, a veces, una discreta prolongación del intervalo PR o defectos de la onda P. Puede haber arritmias auriculares. En los defectos del seno venoso es frecuente hallar un retorno venoso anómalo parcial. Entre sus posibles complicaciones descartan la hipertensión pulmonar en la edad adulta y formación de trombos auriculares, con riesgo de embolias pulmonares, así como eventuales derivaciones derecha-izquierda con posibilidad de embolias sistémicas.
El diagnóstico clínico suele ser evidente y la malformación suele identificarse mediante ecocardiografía bidimensional con flujo Doppler. Sin embargo, a veces se lleva a cabo un cateterismo cardíaco preoperatorio para evaluar el tamaño de la comunicación, para establecer la presencia de venas sistémicas o pulmonares anómalas y para valorar la función del ventrículo izquierdo (VI).
Tratamiento
Se recomienda la corrección quirúrgica programada en los niños de 2 a 6 años de edad, cuando la relación entre el flujo pulmonar y el sistémico es >1,5:1, aunque la mayoría de los pacientes sometidos al cierre de una CIA presentan relaciones 2,5:1 a 3:1. La intervención puede practicarse más antes cuando existen cortocircuitos muy grandes o aparecen arritmias auriculares. En los niños con cortocircuitos pequeños está justificada la observación médica continuada, en ausencia de cardiomegalia o síntomas y si se puede asegurar una asistencia médica constante. Sin embargo, una de las razones aducidas para reparar los defectos pequeños del tabique interauricular es que no es posible predecir qué niños desarrollarán hipertensión pulmonar al llegar a la edad adulta. Otra razón es la prevención de los cortocircuitos derecha-izquierda y las posibles embolias sistémicas.
DEFECTO COMPLETO DEL CANAL AURICULOVENTRICULAR
Defecto de los tabiques auricular y/o ventricular al nivel de las válvulas auriculoventriculares, generalmente asociado a anomalías de las válvulas mitral o tricúspide.
Los defectos del tabique auriculoventricular constituyen aproximadamente el 5% de todas las cardiopatías congénitas. Se dividen en comunicación interauricular tipo ostium primum y defecto completo del canal auriculoventricular (defectode los cojinetes endocárdicos, comunicación auriculoventricular), este último más frecuente en los niños con síndrome de Down.
Los defectos completos del canal auriculoventricular (v. fig. 261-3) pueden producir cianosis desde el nacimiento, debida al cortocircuito auricular o ventricular, posiblemente asociada a soplos sistólicos de insuficiencia mitral o tricúspide. La insuficiencia cardíaca es a menudo precoz, a causa del gran volumen de los cortocircuitos, de la insuficiencia de las válvulas auriculoventriculares o de ambos. Puede dar lugar a una enfermedad pulmonar crónica si la reparación se retrasa más allá de los primeros 6 a 12 meses de vida.
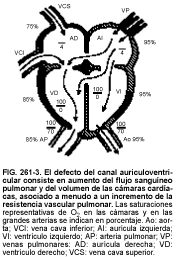
El diagnóstico se establece gracias al patrón del ECG, que muestra desviación del eje hacia la derecha y una rotación en sentido opuesto a las agujas del reloj debida a la ausencia congénita de la rama anterior del fascículo izquierdo, prolongación frecuente del intervalo PR e hipertrofia de VI y VD. La radiografía revela cardiomegalia, con una prominencia típica de la sombra superior de la aurícula derecha, dilatación de VI y VD, tronco de la arteria pulmonar prominente y aumento del flujo sanguíneo arterial pulmonar. La confirmación se obtiene mediante ecocardiografía bidimensional; rara vez son necesarios el cateterismo cardíaco preoperatorio o la angiografía del VI.
La necesidad de corrección quirúrgica depende del estado de salud general del niño. Los defectos completos del canal auriculoventricular deben ser corregidos antes de los 2 años de edad (en muchos centros se intervienen ya a los 3 o 4 meses) para prevenir la enfermedad pulmonar crónica.
DEFECTO PARCIAL DEL CANAL AURICULOVENTRICULAR
(Defecto parcial del tabique auriculoventricular, ostium primum persistente)
Defecto del tabique interauricular inmediatamente por encima de la cresta del tabique interventricular.
En el defecto parcial del canal auriculoventricular se encuentran las mismas anomalías de las válvulas mitral y tricúspide que forman parte del defecto completo. Existen signos clínicos de aumento del flujo sanguíneo pulmonar, con soplo de flujo pulmonar, desdoblamiento persistente de S2 y, a menudo, soplos asociados de insuficiencia mitral o tricuspídea. En el ECG se encuentran un eje superior, una rotación contraria a las agujas del reloj y una sobrecarga de volumen ventricular izquierda o derecha que es proporcional al incremento del flujo sanguíneo pulmonar y a la insuficiencia valvular. La radiografía de tórax revela cardiomegalia, prominencia de la aurícula derecha y del tronco de la arteria pulmonar y aumento de la circulación pulmonar. La información procedente de la ecocardiografía suele ser suficiente para la corrección quirúrgica, por lo que no suele hacerse un cateterismo cardíaco. La intervención se lleva a cabo en la segunda infancia y consiste en la aplicación de un parche, generalmente de pericardio, y, en caso necesario, en la reparación de las válvulas auriculoventriculares.
COMUNICACIÓN INTERVENTRICULAR
Uno o varios orificios en el tabique interventricular que pueden cerrarse espontáneamente durante la lactancia, que pueden provocar una insuficiencia cardíaca, que pueden precisar corrección quirúrgica y que pueden asociarse a enfermedad vascular pulmonar.
Su incidencia global es 2 a 4/1.000 nacidos vivos.
Síntomas, signos y diagnóstico
Las comunicaciones interventriculares pequeñas (CIV, v. fig. 261-4) se manifiestan a menudo por soplos pansistólicos rudos y fuertes que se auscultan en el borde esternal inferior izquierdo durante los primeros meses de la vida y que no se asocian a alteraciones hemodinámicas. Las de mayor tamaño comienzan a auscultarse hacia las 2-3 sem de edad, a medida que la resistencia vascular pulmonar disminuye y la magnitud del cortocircuito izquierda-derecha aumenta. Existe un soplo fuerte, rudo, pansistólico, de grado 3 a 4/6 en el borde esternal inferior izquierdo, junto con un soplo apical mesodiastólico de flujo mitral cuando la comunicación es grande (flujo pulmonar sistémico ³2:1) y el chasquido de cierre pulmonar se acentúa cuando hay aumento de la presión en la arteria pulmonar. La insuficiencia cardíaca, cuando existe, se manifiesta hacia las 6 a 8 sem de edad, a medida que se desarrolla el encharcamiento pulmonar.

Los lactantes con comunicaciones grandes o insuficiencia cardíaca mal controlada corren un riesgo muy alto de sufrir enfermedades graves, con neumonías víricas o bacterianas que a menudo precisan ventilación mecánica. Pueden desarrollar cardiomegalia, insuficiencia cardíaca, retraso del crecimiento o endocarditis infecciosa.
La radiografía revela la presencia de cardiomegalia con dilatación auricular y ventricular izquierda y aumento del flujo sanguíneo pulmonar. Al principio, el ECG refleja la sobrecarga de volumen del VI, pero más tarde, a medida que ascienden las presiones en la arteria pulmonar, muestra también los signos de hipertrofia del VD. No suele ser necesario realizar un cateterismo cardíaco o una angiocardiografía para determinar la localización de la CIV, la resistencia vascular pulmonar o la presencia de anomalías asociadas, a menudo ocultas. La ecocardiografía, con flujo en color y estudios con Doppler, permite obtener una información preoperatoria suficiente.
Tratamiento
La digital, la diuresis y la restricción de sal, junto a la reducción de la poscarga y el tratamiento de las infecciones respiratorias pueden controlar la insuficiencia cardíaca y permitir que el niño tenga un crecimiento y desarrollo satisfactorios. La insuficiencia cardíaca suele desaparecer alrededor del primer o segundo año de vida, a medida que el defecto va siendo menos importante, y puede no precisar corrección quirúrgica.
En los lactantes con insuficiencia cardíaca que responden poco o nada a estas medidas o que tienen cortocircuitos grandes, la corrección quirúrgica durante los primeros meses de vida se plantea cada vez con más frecuencia. Las CIV que siguen siendo grandes y se manifiestan porcardiomegalia, hipocrecimiento o síntomas pero sin insuficiencia cardíaca pueden precisar corrección en etapas posteriores de la infancia. Todos los niños con CIV deben recibir profilaxis contra la endocarditis infecciosa (v. tablas 270-1 y 270-2).
SÍNDROME DEL VENTRÍCULO IZQUIERDO HIPOPLÁSICO
Hipoplasia grave del VI.
La aparición brusca de una insuficiencia cardíaca grave con pérdida de los pulsos periféricos y signos de disminución intensa de la perfusión sistémica en un lactante de 2-3 d previamente sano es muy sugestiva de una atresia mitral o aórtica que depende del conducto arterioso para mantener el flujo y que causa un colapso cardiovascular cuando aquél se cierra. El diagnóstico de sospecha se obtiene ante el hallazgo de cardiomegalia, congestión venosa pulmonar y ausencia de actividad del VI en el ECG, puesta de manifiesto por la falta de la onda Q del tabique o por ondas R precordiales izquierdas positivas. La confirmación procede de la demostración de una hipoplasia grave de las estructuras cardíacas izquierdas en la ecocardiografía. Las intervenciones quirúrgicas más modernas, como el procedimiento de Norwood (conversión en varios tiempos de las cámaras derechas para que funcionen como cámaras izquierdas) son cada vez más efectivas.
TETRALOGÍA DE FALLOT
Anomalía anatómica con obstrucción grave o total del tracto de salida del VD y una CIV que permite a la sangre no oxigenada del VD soslayar la arteria pulmonar y penetrar directamente en el VI y en la aorta.
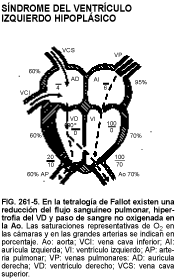
En la tetralogía de Fallot (v. fig. 261-5), se ausculta ya al nacimiento o poco después un soplo de eyección en el borde esternal superior izquierdo, debido a la obstrucción del tracto de salida del VD, seguido por la aparición progresiva de cianosis. No obstante, los RN que sufren una atresia de la válvula pulmonar o cuyo flujo sanguíneo pulmonar depende enteramente del conducto arterioso presentan una cianosis intensa y un soplo continuo de flujo ductal. Los lactantes mayores tienen un soplo de eyección con desviación del eje hacia la derecha e hipertrofia del VD en el ECG; la radiografía muestra un corazón pequeño y un tronco de la arteria pulmonar cóncavo, con una circulación pulmonar reducida. En el 25% de estos niños se encuentra un arco aórtico derecho. Algunos RN y lactantes desarrollan crisis hipercianóticas con ansiedad, signos de ahogo, sufrimiento respiratorio y alteración de la conciencia, generalmente asociadas a la actividad.
En los lactantes con cianosis por cierre del conducto arterioso, la infusión de prostaglandinaE1 en dosis de 0,05 a 0,1 mg/kg/min i.v. suele bastar para mantener la permeabilidad ductal hasta que pueda llevarse a cabo un procedimiento paliativo, consistente en una anastomosis sistémica-pulmonar (anastomosis de las arterias subclavia y pulmonar, o sus modificaciones). La infusión de prostaglandina puede provocar una parada respiratoria, por lo que debe disponerse de un equipo de reanimación (v. Reanimación cardiopulmonar, cap. 263). La cantidad administrada debe reducirse a la dosis eficaz más baja lo antes posible.
El tratamiento de las crisis hipercianóticas consiste en suplementos de O2, colocación del niño en posición genupectoral y administración de morfina en dosis de 0,1 a 0,2 mg/kg i.m. El uso de propranolol en dosis de 0,25 a 1,0 mg/kg v.o. cada 6 h puede prevenir la aparición de crisis posteriores, pero el cateterismo, la angiocardiografía y la corrección o paliación quirúrgica deben ser considerados medidas urgentes. La reparación precoz del VD es el procedimiento que mejor protege la función del VD y el que tiene mejores resultados. Algunos lactantes precisan incrementos paliativos del flujo sanguíneo pulmonar, que se consiguen mediante anastomosis entre las circulaciones sistémica y pulmonar.
TRASPOSICIÓN DE LAS GRANDES ARTERIAS
Anomalía anatómica en la que la aorta nace directamente del VD y la arteria pulmonar surge del VI, provocando una hipoxemia sistémica grave.
La trasposición de las grandes arterias constituye el 5 al 7% de todas las cardiopatías congénitas.
Los lactantes con esta cardiopatía (v. fig. 261-6) suelen estar sanos por lo demás pero presentan, inmediatamente después del nacimiento, una cianosis intensa que progresa con rapidez hacia una acidosis metabólica, causada por la deficiente oxigenación de los tejidos, con alcalosis respiratoria compensadora. La radiología de tórax revela una base ancha por la superposición de los grandes vasos, que no se localizan en paralelo; el segmento principal de la arteria pulmonar no se encuentra en su localización habitual, y el corazón presenta la morfología ovoide. La congestión venosa pulmonar se desarrolla rápidamente. El ECG del RN es normal.

El diagnóstico debe confirmarse de inmediato mediante la ecocardiografía. Estos pacientes son candidatos a la intervención paliativa, consistente en septostomía con balón para mejorar la mezcla auricular y descomprimir la aurícula izquierda. Los lactantes en estado crítico con hipoxemia pueden recibir prostaglandina E1, que produce una apertura del conducto arterioso, incrementa el flujo sanguíneo pulmonar y mejora transitoriamente la oxigenación sistémica, aunque también puede ser necesaria la septostomía auricular inmediata. La corrección quirúrgica mediante una técnica de intercambio vascular con reimplantación de las arterias coronarias se lleva a cabo en los primeros 7 a 10 d de vida.
CARDIOPATÍA CONGÉNITA CIANÓTICA COMPLEJA
Anomalías cardíacas complejas productoras de cianosis.
Las anomalías más complejas (p. ej., ventrículo único con o sin estenosis pulmonar, atresia tricúspide con o sin trasposición de las grandes arterias, atresia tricúspide y pulmonar, atresia mitral, tronco arterioso) son menos frecuentes. Las técnicas de imagen habituales suelen permitir su diagnóstico anatómico, que debe ser confirmado mediante angiografía.
El tratamiento inicial suele consistir en asegurar un riego sanguíneo pulmonar suficiente mediante anastomosis entre los vasos sistémicos y pulmonares o en proteger el lecho vascular pulmonar y controlar el aumento de flujo en el mismo mediante el cerclaje de la arteria pulmonar. En algunos niños puede restablecerse un patrón de flujo normal y separar las sangres oxigenada y desoxigenada con una técnica de Fontan modificada, que dirige el flujo sanguíneo desde la aurícula derecha hacia la arteria pulmonar, excluyendo al ventrículo de la circulación derecha. A veces es también posible reparar el tronco arterioso, separando la arteria pulmonar del tronco común e insertando un conducto provisto de válvulas para dirigir la sangre desde el ventrículo derecho hacia el árbol arterial pulmonar.
ESTENOSIS DE LA VÁLVULA AÓRTICA
Estrechamiento del orificio de la válvula aórtica debido a una anomalía congénita de la misma, habitualmente una válvula bicúspide
La estenosis valvular aórtica constituye alrededor del 5% de todas las malformaciones cardíacas con manifestaciones clínicas, pero la verdadera prevalencia de la válvula aórtica bicúspide no diagnosticada es, probablemente, mucho mayor; quizá sea, incluso, la cardiopatía congénita más frecuente.
La obstrucción puede ser valvular, subvalvular (subaórtica) o supravalvular. Esta última es rara, pero a menudo va acompañada de hipercalcemia (síndrome de Williams) y puede asociarse a la estenosis periférica de la arteria pulmonar. Los síntomas principales son los de la enfermedad subyacente, si bien las obstrucciones graves pueden producir dolor torácico, cuadros de tipo sincopal o síncope con actividad.
A veces, las válvulas aórticas bicúspides congénitas producen cuadros de obstrucción grave durante la niñez, pero es más frecuente que la obstrucción se manifieste durante la vida adulta.
Síntomas, signos y diagnóstico
En raras ocasiones, la estenosis valvular sintomática se manifiesta durante la lactancia y provoca una obstrucción grave al flujo de salida del VI con disfunción ventricular grave, insuficienciacardíaca, cambios isquémicos (visibles en el ECG) y un escaso gasto cardíaco sistémico. Con más frecuencia se manifiesta en etapas posteriores de la infancia en forma de válvula aórtica bicúspide con estenosis y, a veces, con insuficiencia. En general no produce síntomas. Sus signos consisten en un soplo sistólico de eyección, más fuerte en el borde esternal superior derecho, generalmente asociado a un claro chasquido sistólico de eyección y una acentuación del ruido de cierre de la válvula aórtica. También puede auscultarse un soplo protodiastólico suave de insuficiencia aórtica. La endocarditis o endarteritis infecciosas son complicaciones de la válvula aórtica bicúspide.
La obstrucción subvalvular debida a un caballete fibroso o a una hipertrofia muscular produce prácticamente los mismos signos, excepto que no se ausculta el chasquido de eyección, que el soplo diastólico puede ser más intenso en la parte media del esternón y que no existe dilatación postestenótica. Suele ser asintomática.
El ECG muestra signos progresivos de hipertrofia del VI y de isquemia, a medida que la función del ventrículo se deteriora, pero estos hallazgos no guardan correlación con el grado de obstrucción. La radiografía puede demostrar la dilatación postestenótica de la aorta ascendente y, en las obstrucciones de larga duración, la hipertrofia del VI. Puede ser necesario efectuar una evaluación mediante ecocardiografía, cateterismo cardíaco y angiocardiografía, en los pacientes con obstrucción grave o con sintomatología (p.ej., dolor torácico de esfuerzo, síncope).
Tratamiento
Durante la lactancia, la insuficiencia cardíaca con peligro para la vida obliga a la valvulotomía aórtica inmediata para evitar la muerte, bien por valvuloplastia con balón, bien mediante valvulotomía quirúrgica. Los resultados no son siempre buenos y a veces persiste una insuficiencia valvular residual. Cuando el niño crece, suele ser necesario realizar una sustitución valvular, que se lleva a cabo hacia la mitad o el final de la infancia, aunque a veces puede posponerse hasta la edad adulta.
La paliación mediante valvulotomía aórtica está indicada cuando existen, al mismo tiempo, un gradiente VI-aorta importante (³50 mm Hg) y signos y síntomas de disfunción del VI. Se trata de un procedimiento paliativo más que curativo, y a largo plazo son frecuentes las reestenosis, que obligan a la sustitución valvular. También una insuficiencia aórtica postoperatoria importante, puede requerir la sustitución. Los pacientes con válvulas aórticas bicúspides congénitas que presentan obstrucción durante la edad adulta también suelen precisar prótesis valvulares. La reparación quirúrgica de la obstrucción aórtica supravalvular, aunque posible, es difícil. Para corregir la obstrucción subvalvular debida a un caballete fibroso, debe extirparse por completo el tejido anómalo que, sin embargo, suele volver a crecer, provocando una obstrucción y obligando a repetir la resección. La estenosis subaórtica secundaria a una cardiomiopatía puede tratarse con bloqueo ß, miotomía o miomectomía.
En todo paciente con obstrucción al tracto de salida del ventrículo izquierdo, cualquiera que sea su nivel, está indicada la profilaxis antibiótica contra la endocarditis o endarteritis infecciosa (v.tablas 270-1 y 270-2).
ESTENOSIS DE LA VÁLVULA PULMONAR
Estrechamiento del orificio de la válvula pulmonar debida a una anomalía congénita de la misma, siendo la más frecuente la válvula en cúpula, a veces bicúspide, con limitación de la apertura.
La estenosis de la válvula pulmonar, de grado variable, constituye alrededor del 10% de todas las cardiopatías congénitas.
Síntomas, signos y diagnóstico
En el período neonatal, la estenosis de la válvula pulmonar con obstrucción grave al flujo de salida del VD puede constituir una urgencia médica, con cortocircuito auricular derecha-izquierda, y exige un diagnóstico inmediato: el ECG demuestra una disminución de las fuerzas del VD (cuando el VD es hipoplásico, trastorno generalmente grave) o una hipertrofia del mismo; en la radiografía se observa un flujo sanguíneo pulmonar muy reducido; la ecocardiografía revela una válvula muy estrecha con escasos movimientos. También son útiles el cateterismo cardíaco y la angiografía para valorar la función potencial del VD.
En los niños mayores no suele encontrarse cianosis, pero muchos de ellos presentan defectos de la coloración periférica debidos al largo tiempo decirculación y a la gran diferencia arteriovenosade O2. Existe un soplo de eyección, habitualmente asociado a un chasquido prominente, que se ausculta en el borde esternal superior izquierdo y que presenta un máximo progresivamente tardío, a medida que la obstrucción avanza. El componente pulmonar de S2 se retrasa y disminuye cada vez más. El ECG demuestra una hipertrofia creciente del VD. En la radiografía se observan un corazón de tamaño normal con prominencia creciente del tronco de la pulmonar, que aumenta al agravarse la estenosis. Las marcas de la circulación pulmonar son cada vez más estrechas.
Tratamiento
En el recién nacido es necesario crear inmediatamente una anastomosis sistémica-pulmonar (p. ej., un procedimiento de Blalock-Taussig) para proporcionar un flujo pulmonar adecuado. Hasta entonces, puede obtenerse una paliación transitoria manteniendo abierto el conducto arterioso con prostaglandina E1 (alprostadil), en dosis de 0,05 a 0,1 mg/kg/min i.v., para salvar la vida del niño. También la valvulotomía pulmonar puede estar indicada. Estos procedimientos son paliativos; en fases posteriores de la infancia pueden ser necesarios otros procedimientos quirúrgicos para mejorar el gasto del VD. La valvuloplastia con balón puede ser útil en los lactantes con VD normales o casi normales.
En niños mayores, el procedimiento más utilizado es la valvuloplastia con balón, asociando un cateterismo para valorar el estado del tabique interauricular y obtener determinaciones exactas de las presiones en la válvula pulmonar. Algunas válvulas muy displásicas obligan a la valvulotomía quirúrgica. Cuando la estenosis no es grave, el tratamiento puede retrasarse hasta inmediatamente antes de la edad preescolar.
ESTENOSIS PULMONAR PERIFÉRICA
Zonas múltiples de estenosis luminal en las ramas de la arteria pulmonar.
Muchos RN presentan soplos sistólicos suaves en las ramas de la arteria pulmonar, sin signos ni síntomas de aumento significativo de las presiones cardíacas derechas. Estos soplos suelen desaparecer después del primer año, a medida que el lactante crece. Sin embargo, en los niños que padecen ciertos procesos (p. ej., rubéola congénita, síndrome de hipercalcemia [síndrome de Williams]) y en algunos niños sanos por lo demás, existe una obstrucción anatómica al flujo en alguna de las ramas de la arteria pulmonar, que produce un soplo continuo. Esta obstrucción no suele ser susceptible de corrección quirúrgica, debido a que existen múltiples áreas de estenosis en el interior del pulmón y porque el árbol arterial distal a la obstrucción suele ser hipoplásico. En algunos casos puede ser útil la dilatación con vástagos.
PERSISTENCIA DEL CONDUCTO ARTERIOSO
Fracaso del cierre del conducto que comunica la arteria pulmonar y la aorta.
La persistencia del conducto arterioso es común en los prematuros y se encuentra en hasta un 80% de todos los niños nacidos antes de las 28sem. Esta frecuencia disminuye progresivamente al aumentar la edad gestacional. La permeabilidad persistente del conducto arterioso en el niño a término afecta a 1 de cada 2.000 nacidos vivos.
Síntomas, signos y diagnóstico
En el niño prematuro, la persistencia del conducto arterioso (PCA, v. fig. 261-7) suele asociarse a un aumento del flujo sanguíneo pulmonar y compromete aún más el intercambio de gases, sobre todo en los que padecen un síndrome de sufrimiento respiratorio. El niño presenta pulsos saltones, un precordio hiperdinámico y un aumento del ruido de cierre de la válvula pulmonar. Típicamente se ausculta un soplo en el área pulmonar que puede ser continuo, sistólico con un pequeño componente diastólico o sólo sistólico. Este soplo falta en algunos niños que, sin embargo, presentan los signos restantes. El ECG suele corresponder al grado de prematuridad (prominencia del VI), pero la carga de volumen del VI puede ser mayor de lo normal. En la radiografía se encuentran cardiomegalia y, si los cambios pulmonares del sufrimiento respiratorio no son intensos, un aumento del flujo sanguíneo pulmonar. La ecocardiografía puede revelar un diámetro auricular izquierdo superior al diámetro de la raíz de la aorta. En la ecocardiografía con Doppler color suelen observase una inversión del flujo en la arteria pulmonar durante la diástole o imágenes de la totalidad del conducto arterioso.

En el niño a término, la PCA suele diagnosticarse después de las 6 a 8 sem de vida, al auscultar un soplo continuo en el borde esternal superior izquierdo. Los pulsos periféricos son llenos, con aumento de la presión diferencial. El ECG puede revelar la sobrecarga de volumen del VI. En la radiografía se observan la prominencia de la aurícula izquierda, del VI y de la aorta ascendente, así como el aumento del riego sanguíneo pulmonar, si el conducto arterioso mantiene un flujo importante. Es preciso comprobar que la permeabilidad del conducto arterioso no está compensando una atresia pulmonar y que el soplo no corresponde a una fístula arteriovenosa sistémica, una estenosis de las ramas de la arteria pulmonar o una ventana aortopulmonar. También es necesario evaluar los pulsos femorales y la PA de las piernas para descartar una coartación oculta.
Tratamiento
En los niños prematuros con síndrome de distrés (sufrimiento) respiratorio, debe intentarse el cierre del conducto arterioso mediante restricción de líquidos (90 a 100 ml/kg/d), diuresis, mantenimiento de una buena oxigenación, fármacos (p. ej., indometacina) o ligadura quirúrgica. La restricción moderada de líquidos durante 2 a 3 d debe ir seguida de un aporte progresivo y constante. En ausencia de ictericia excesiva (bilirrubina indirecta >10 mg/dl [>170 m mol/l]), insuficiencia renal (creatinina >1,4 mg/dl [>120m mol/l], BUN >35 mg/dl [>12,5 mmol urea/l]) o trombocitopenia (recuento plaquetario <100.000/ml), tras la indometacina, 1 o 2 dosis de 0,2 mg/kg i.v. administradas con 12 h de diferencia, permite conseguir rápidamente el cierre del conducto. Aunque éste suele cerrarse en los prematuros bien espontáneamente, bien con restricción de líquidos tan sólo, cuando sigue siendo permeable precisa una ligadura quirúrgica, que se efectúa entre los 11/2 y 21/2 años de edad.
En los nacidos a término, la ligadura quirúrgica o la sección están indicadas en caso de insuficiencia cardíaca. Entre los 6 meses y los 3 años de edad puede llevarse a cabo una intervención programada para prevenir el desarrollo de una endarteritis infecciosa.
COARTACIÓN DE LA AORTA
Estrechamiento localizado de la luz de la aorta.
La coartación de la aorta supone el 7-8% de todas las cardiopatías congénitas.
Síntomas, signos y diagnóstico
Los lactantes con coartación de la aorta pueden presentar una insuficiencia cardíaca de comienzo brusco, colapso cardiovascular y acidosis metabólica grave cuando el conducto arterioso se cierra y se compromete la circulación distal. En los niños mayores, se encuentra una hipertensión relativa de los miembros superiores en comparación con los inferiores y también pueden tener una hipertensión absoluta. Sobre la coartación se ausculta a menudo un soplo suave, que se oye en el área pulmonar pero que suele ser más fuerte enla espalda. Los pulsos femorales, aunque a menudo palpables, suelen ser menores y más tardíos que los braquiales. La radiografía puede revelar la presencia de muescas en las costillas, debidas al aumento del flujo sanguíneo y consiguiente dilatación de las arterias mamarias internas, aunque en general no son visibles antes de los 10 a 12 años.
En todos los niños, las arterias colaterales del reborde escapular están dilatadas y a menudo son palpables. El ECG suele ser normal pero puede mostrar hipertrofia del VI. Las radiografías revelan un corazón de tamaño normal, pero la coartación puede verse cuando se obtiene una proyección ligeramente oblicua anterior izquierda. No es necesario practicar un cateterismo ni una angiografía, salvo que existan otros defectos asociados importantes (p. ej., estenosis aórtica, insuficiencia aórtica, valvulopatía mitral o comunicación interventricular) o se sospeche que el segmento estenótico no se halla en su localización normal, inmediatamente distal a la arteria subclavia izquierda, o que es más largo de lo habitual.
Tratamiento
En los lactantes debe hacerse un tratamiento de sostén farmacológico inmediato (p. ej., dobutamina, 5 a 15 mg/kg/min, furosemida 1 a 2 mg/kg. i.v. ycon menos frecuencia adrenalina), intubación y asistencia respiratoria si procede y perfusión de prostaglandina E1 en dosis de 0,05 a 0,1 mg/kg/min i.v. para reabrir el conducto arterioso. La respuesta a la administración de prostaglandina es gradual y suele producirse a lo largo de varias horas, pero casi todos los lactantes se estabilizan, su perfusión distal aumenta y el riego sanguíneo renal mejora, al tiempo se resuelve la acidosis metabólica, lo que permite la corrección quirúrgica de la coartación y el cierre del conducto arterioso permeable.
En los niños mayores, se recomienda hacer la corrección quirúrgica, consistente en la extirpación y anastomosis directa, técnicas de «techado» con la arteria subclavia o colocación de injertos en caso necesario, hacia los 4 a 6 años de edad o antes, en caso de hipertensión de las extremidades superiores, insuficiencia cardíaca u otras complicaciones. También es necesaria la profilaxis contra la endocarditis (v. tablas 270-1 y 270-2).
TRONCO ARTERIOSO PERSISTENTE
Malformación en la cual el tronco primitivo no se divide en dos vasos independientes, por lo que las arterias pulmonares nacen de la aorta y no del ventrículo derecho.
Los signos clínicos suelen aparecer algunos días o semanas después del nacimiento y consisten en las manifestaciones de la insuficiencia cardíaca y una ligera insaturación. Son característicos el precordio hiperdinámico, el aumento de la presión del pulso, un S1 normal, con frecuente chasquido de eyección, y un S2 alto, generalmente único. Los soplos cardíacos son variables y pueden comprender un soplo de flujo en la base, un soplo alto de insuficiencia en el borde esternal inferior izquierdo y un soplo mesosistólico mitral. Cuando las válvulas del tronco son insuficientes, se ausculta un soplo diastólico de tono alto al nivel de la parte media del esternón. El ECG suele mostrar una hipertrofia biventricular. En la radiografía de tórax se encuentran cardiomegalia, aumento del flujo sanguíneo pulmonar, un arco aórtico derecho (en alrededor de 1/3 de los casos) y unas arterias pulmonares en posición relativamente alta. La ecocardiografía permite confirmar el diagnóstico y habitualmente se requieren también un cateterismo y una angiografía para planificar la intervención quirúrgica. El tratamiento es necesario para evitar la insuficiencia cardíaca y está indicada la corrección quirúrgica precoz, con colocación de un conducto provisto de válvulas entre el ventrículo derecho y las arterias pulmonares, que se extirpan del tronco arterioso. Esta reparación debe hacerse lo antes posible para prevenir la enfermedad vascular pulmonar.
ANOMALÍAS MENOS FRECUENTES
Las anomalías menos frecuentes (p. ej., malformación de Ebstein de la válvula tricúspide), la presencia de afectación cardíaca en las enfermedades fetales (p. ej., anemia intrauterina prolongada, arritmias fetales), las malformaciones cardíacas graves asociadas al síndrome de asplenia y la disfunción cardíaca secundaria a causas no cardíacas (p. ej., hipotiroidismo) precisan un tratamiento individualizado.
Muchas alteraciones raras (p. ej., bloqueo cardíaco completo congénito, errores congénitos del metabolismo que suelen producir acidosis grave y disfunción miocárdica secundaria, síndrome de prolongación de QT con riesgo de arritmia grave posiblemente mortal) y las malformaciones poco comunes (p. ej., cor triatriatum) precisan medidas diagnósticas y terapéuticas especiales.
ENFERMEDAD VASCULAR PULMONAR
(Reacción de Eisenmenger)
Hipertensión pulmonar secundaria a las alteraciones de la circulación pulmonar que se producen en las cardiopatías congénitas de larga duración.
La enfermedad vascular pulmonar puede ser un factor que dificulte la asistencia prestada a los lactantes y niños con cardiopatías congénitas (p.ej., comunicación interventricular, defectos del canal auriculoventricular, tronco arterioso) que dan lugar a cortocircuitos de alta presión (ventriculares, aortopulmonares) y puede ser un trastorno que aparezca por sí solo, como hipertensión pulmonar primaria. En estos casos, la resistencia vascular pulmonar se mantiene elevada, con hipertrofia muscular de la capa media de las arteriolas pulmonares y oclusión de muchas de las ramas más pequeñas.
A medida que la resistencia vascular pulmonar se aproxima y alcanza a la resistencia vascular sistémica, los cortocircuitos izquierda-derecha disminuyen, mientras que los de sentido derecha-izquierda provocan una desaturación sistémica cada vez mayor y una cianosis visible. La persistencia y el aumento de los cortocircuitos derecha-izquierda causan una hipoxemia progresiva, con una policitemia creciente. También aumenta la capacidad de transporte de O2, como demuestra el incremento del Hto a 65% o más, pero, con valores muy altos, la gran viscosidad de la sangre dificulta la liberación de O2 a los tejidos.
La cirugía suele estar contraindicada cuando se calcula que la resistencia vascular pulmonar es >1/2 de la resistencia vascular sistémica calculada. La posibilidad de corrección debe ser valorada con gran cuidado siempre que la resistencia vascular pulmonar alcance cifras superiores a las normales, sean las que sean. En estos casos, una flebotomía prudente, manteniendo el volumen sanguíneo total, puede ser beneficiosa, pues reducirá los valores de Hto desde >65% hasta alrededor del 60% en los pacientes sintomáticos (p. ej., los que tienen dificultades para hablar, problemas visuales o fatiga intensa).
No existe tratamiento específico para la enfermedad vascular pulmonar, aunque en la actualidad se está investigando el uso de agentes tales como la prostaciclina.
INSUFICIENCIA CARDÍACA
Síndrome clínico que se produce cuando el corazón no puede mantener un gasto cardíaco suficiente para satisfacer las necesidades metabólicas del organismo, incluyendo las que sustentan el crecimiento.
Las causas de la insuficiencia cardíaca (IC) se enumeran en la tabla 261-1. Entre las causas no cardíacas se encuentran la anemia crónica, la obstrucción de las vías respiratorias altas, las deficiencias nutricionales, la asfixia, la toxicidad medicamentosa (p. ej., por daunorubicina) y algunas enfermedades sistémicas (p. ej., enfermedades por depósito, ataxia de Friedreich y hemodilución yatrógena). El síndrome del ventrículo izquierdo hipoplásico suele manifestarse entre 48 y 72 h después del nacimiento por una IC repentina y una acidosis metabólica que se debe a la escasa perfusión sistémica.

Síntomas, signos y pruebas analíticas
En los lactantes, el comienzo de la IC puede ser gradual pero suele ser rápido y, en ocasiones, sumamente rápido. Casi siempre se encuentra taquicardia, con frecuencias cardíacas >120-140 latidos/min y hasta 200 latidos/min. En este grupo de edad suele haber signos de IC derecha e izquierda simultáneamente.
La insuficiencia del VI se manifiesta con síntomas de dificultad respiratoria. La congestión venosa pulmonar, el aumento de la presión capilar pulmonar y el paso de líquido a los alvéolos, al intersticio y a los espacios bronquiolares producen disnea y taquipnea, con frecuencias respiratorias >60 a 100 respiraciones/min, en ausencia de enfermedad pulmonar primaria. Una infección sobreañadida puede agravar el cuadro. Son frecuentes la tos y las sibilancias; los estertores y los roncus son variables, pero no raros, mientras que el edema pulmonar franco, con expectoración espumosa y sanguinolenta sí lo es. El incremento de la frecuencia respiratoria produce fatiga y aumento de las demandas metabólicas. Todo ello conduce al rechazo del alimento, a una ingesta insuficiente y al retraso del crecimiento aunque, por lo general, el perímetro cefálico y el crecimiento longitudinal se mantienen. El retraso del crecimiento puede quedar enmascarado en parte por la retención de líquido y el menor volumen urinario, que producen una falsa ganancia ponderal. Otros síntomas son inquietud, irritabilidad y sudoración excesiva.
La cardiomegalia es común, salvo en los casos de pericarditis constrictiva y de obstrucción venosa pulmonar grave. La deficiente función del miocardio se refleja en la debilidad de los ruidos cardíacos, los signos de escasa perfusión periférica con extremidades frías y en los menores volumen del pulso y llenado capilar, junto a un color grisáceo, más que azul, de la piel. La cianosis, un signo de cortocircuito derecha-izquierda intracardíaco, puede indicar un defecto del intercambio gaseoso alveolar secundario a la congestión venosa pulmonar o al bajo gasto cardíaco con incremento de la diferencia arteriovenosa de O2.
En la insuficiencia del VD, el signo más frecuente y fidedigno durante la lactancia es la hepatomegalia, que también es un indicador sensible de la eficacia del tratamiento. El dolor espontáneo y a la palpación debido a la congestión hepática y las alteraciones del pulso venoso yugular, aunque útiles en los niños mayores, no son fiables en el lactante. A veces se encuentran edemas periféricos, sobre todo en el dorso de las manos y de los pies y en la región periorbitaria.
Pocos datos analíticos son específicos de la IC. Pueden encontrarse una anemia por dilución y una hipernatremia. También puede haber disminución del volumen urinario y albuminuria. La hipoglucemia, secundaria al agotamiento de unos depósitos insuficientes de glucógeno y al estado hipermetabólico, es frecuente. El recuento leucocitario puede indicar una infección asociada y la prolongada desaturación arterial sistémica suele inducir el desarrollo de policitemia y, más tarde, de anemia ferropénica.
Diagnóstico
La IC se diagnostica por sus síntomas y signos, que reflejan la congestión pulmonar o sistémica aguda o crónica, y por los de la alteración cardíaca subyacente; por ejemplo, la estenosis pulmonar puede provocar una IC sin congestión pulmonar. Debe intentarse el diagnóstico anatómico específico mediante la anamnesis, la exploración física y los hallazgos radiológicos y analíticos básicos.
Es preciso palpar el precordio para buscar frémitos, desplazamientos de la punta y localización del impulso máximo. Hay que valorar la calidad e intensidad de los ruidos cardíacos, auscultando el cierre de las dos válvulas semilunares y su sincronización relativa, así como los ruidos adicionales. Los soplos deben identificarse según su localización, momento de aparición, duración, intensidad y calidad. Deben explorarse los pulmones para evaluar la presencia de congestión o infección. También hay que valorar la calidad de los pulsos periféricos y medir la PA en todos los miembros. El grado de desaturación periférica de O2 y de anemia puede determinarse examinando las conjuntivas, las membranas mucosas, los labios y los lechos ungueales. Deben registrarse el tamaño y firmeza del hígado y los edemas periféricos. Para valorar la retención de líquidos, lo mejor es registrar cuidadosamente los incrementos sucesivos de peso. Una reevaluación frecuente de estos hallazgos físicos permitirá establecer la eficacia del tratamiento y ayudará a hacer el diagnóstico específico.
El ECG es de escasa ayuda en el diagnóstico de la IC, pero tiene valor para llegar al diagnóstico anatómico específico. La ecocardiografía de flujo en color, cartografía y estudios Doppler, el cateterismo cardíaco y la angiocardiografía no son de utilidad para diagnosticar la IC, pero a veces resultan fundamentales para establecer el diagnóstico anatómico exacto. Ninguna de estas pruebas suele llevarse a cabo antes de controlar la IC y otros problemas agudos (p. ej., alteraciones de los electrolitos, infecciones).
Pronóstico y tratamiento
El pronóstico depende fundamentalmente de la enfermedad subyacente y de su tratamiento.
Algunos casos de IC intrauterina pueden tratarse corrigiendo la enfermedad subyacente, por ejemplo, la digitalización y la diuresis maternas pueden corregir una taquicardia fetal.
El tratamiento de la IC incluye O2 humidificado administrado mediante tienda, mascarilla osonda nasal, con un O2 inspirado adecuado (<40% para evitar la lesión del epitelio pulmonar), a fin de prevenir la cianosis y aliviar el sufrimiento respiratorio; sedación con sulfato de morfina en dosis de 0,2 mg/kg s.c. cada 4 a 6 h según las necesidades, y elevación de la cabeza. Si bien los niños mayores pueden beneficiarse de la posición en «silla cardíaca» (sentados, con el tronco semierecto y las piernas elevadas), esta posición puede agravar el compromiso respiratorio de los lactantes, ya que las vísceras abdominales ascienden hacia el tórax. Debe limitarse el aporte de Na y, en menor medida, restringir los líquidos hasta niveles de mantenimiento, lo que ayuda a mantener una respuesta favorable al tratamiento, aunque conviene evitar los niveles séricos de Na <130 mEq/l. Los torniquetes rotatorios, la flebotomía y la asistencia respiratoria son usados con menos frecuencia. Son de gran valor otras medidas generales de sostén (p. ej., uso de fórmulas con mayor densidad calórica, control riguroso de la fiebre, tratamiento de la anemia).
La digoxina sigue siendo un fármaco muy utilizado en el tratamiento de la IC (v. tabla 261-2). La dosis de digitalización inicial puede administrarse i.v. o v.o. en 3 tomas divididas, con una porción inicial más grande o cada 4, 6 u 8 h, dependiendo de la urgencia. La digitoxina i.m. no suele estar indicada. El mantenimiento con dos dosis diarias suele lograr una respuesta más suave que la administración de una sola dosis diaria. Es importante extremar las precauciones al prescribir digoxina. Este fármaco se presenta con una concentración de 50 mg/ml (0,05 mg/ml) en el elixir para uso oral y de 250 mg/ml (0,25 mg/ml) en las ampollas para uso i.v. La medición de sus niveles sanguíneos en los RN y lactantes no es ni fidedigna ni muy útil.

La diuresis con furosemida o ácido etacrínico en dosis de 1 mg/kg i.v. o 2 mg/kg v.o. produce una respuesta inmediata. La administración de ambos fármacos puede repetirse cada 4 a 6 h, duplicando la dosis si es necesario. Para el tratamiento diurético a largo plazo de los lactantes y niños puede emplearse la clorotiazida en dosis de 20 a 40 mg/kg/d v.o., dividida en dos tomas. La interrupción del tratamiento (p. ej., durante 3 o 4 d/sem) ayuda a prevenir los desequilibrios electrolíticos, pero los suplementos de K pueden ser necesarios. Los diuréticos deben prescribirse con precaución en caso de patología renal aguda o crónica.
En los casos de IC muy grave en los que no es posible mejorar el gasto cardíaco con ninguna otra medida, pueden ser útiles la dopamina o la dobutamina en dosis de 5 mg/kg/min (aumentando a 15 mg/kg/min, si es necesario). Conviene evitar las dosis más altas por el riesgo de efectos adversos para el flujo sanguíneo renal. La reducción de la poscarga se consigue con nitroprusiatoen dosis de 0,5 a 3,0 mg/kg/min i.v., hidralazina en dosis de 0,5 a 5,0 mg/kg/d oral (en 2 a 4 tomas divididas, hasta una dosis máxima de 7,5 mg/kg/d en los niños mayores y de 5 mg/kg/d en los lactantes) y captoprilo en dosis de 0,5 a 6,0 mg/kg/d v.o. (2 a 4 tomas), aunque todos estos fármacos deben usarse con precaución. Otros agentes, como la amrinona, se reservan para la IC grave y sólo se administran en el marco de los cuidados intensivos.
DEFECTOS GASTROINTESTINALES
Los RN con obstrucción intestinal congénita presentan distensión y vómitos ya al nacimiento o en los 1 a 2 d siguientes. El tratamiento inmediato consiste en descomprimir el abdomen con aspiración nasogástrica continua para evitar la emesis, que puede provocar una neumonía aspirativa o incrementar más aún la distensión abdominal, con el consiguiente compromiso respiratorio, y remisión a un centro donde se practique la cirugía neonatal. También es fundamental mantener la temperatura corporal, prevenir la hipoglucemia con dextrosa al 10% y electrolitos i.v. y evitar o corregir la acidosis y las infecciones, para que el niño llegue al quirófano en condiciones óptimas. Cuando un lactante presenta una malformación congénita, debe investigarse la presencia de anomalías adicionales en otras vísceras, sobre todo en el SNC, el corazón y los riñones.
OBSTRUCCIÓN GASTROINTESTINAL ALTA
Debe sospecharse una obstrucción gastrointestinal alta (esofágica, gástrica, duodenal o yeyunal) en todos los casos de hidramnios (exceso de volumen del líquido amniótico). El hidramnios se produce cuando el feto no puede tragar y absorber el líquido amniótico o en casos de emesis fetal. Es preciso introducir una sonda nasogástrica en el estómago del RN inmediatamente después del parto; el hallazgo de >30 ml de líquido en su interior, especialmente si está teñido con bilis, respalda el diagnóstico de obstrucción GI alta, mientras que la imposibilidad de hacer progresar la sonda sugiere una atresia esofágica.
Atresia esofágica
La atresia esofágica se asocia a fístula traqueoesofágica, casi siempre de tipo III B (fístula que se extiende desde la vecindad de la carina traqueal hasta el tercio inferior del esófago, v. fig. 261-8), en el 80% de los casos. El cuadro característico consta de secreciones excesivas, tos y cianosis con los intentos de alimentación, y neumonía aspirativa. Las lesiones de tipo III B producen rápidamente distensión abdominal, pues, al llorar el niño, el aire de la tráquea penetra en el esófago inferior y el estómago a través de la fístula.
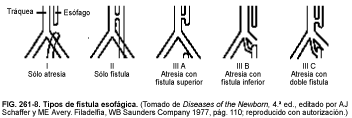
Diagnóstico
El diagnóstico se sospecha por la imposibilidad de hacer progresar la sonda nasogástrica hasta el estómago; con rayos X, una sonda radioopaca permite establecer la localización de la atresia. En los casos atípicos puede ser necesario introducir un poco de contraste radiológico hidrosoluble en la bolsa superior del esófago bajo control radioscópico para determinar la anatomía: el medio de contraste debe ser aspirado nuevamente con rapidez, ya que su paso a los pulmones puede provocar una neumonitis química. El procedimiento sólo debe ser llevado a cabo por un radiólogo experto en un centro que disponga de cirugía neonatal.
Tratamiento
El propósito del tratamiento preoperatorio es conseguir que el niño se encuentre en condiciones óptimas antes de la cirugía y en prevenir la neumonía aspirativa, que hace más peligrosa la intervención quirúrgica. Debe suprimirse la alimentación oral. La aspiración continua con una sonda de aspiración de doble luz situada en la bolsa esofágica superior previene la aspiración de la saliva deglutida. El lactante debe colocarse en decúbito prono con la cabeza elevada 30-40° y acostado sobre el lado derecho para facilitar el vaciado gástrico y minimizar el riesgo de aspiración de jugo gástrico a través de la fístula. Si la corrección quirúrgica definitiva debe demorarse a causa de una prematuridad extrema, de una neumonía aspirativa o de las malformaciones congénitas asociadas, se hará una gastrostomía para descomprimir el estómago. La aspiración a través de la sonda de gastrostomía reduce el riesgo de reflujo del contenido gástrico al árbol traqueobronquial. Cuando el RN se estabiliza, se lleva a cabo la corrección quirúrgica extrapleural de la fístula. En ocasiones, la distancia entre los dos segmentos esofágicos es demasiado grande y no permite la corrección primaria. En estos casos, pueden resultar útiles el estiramiento suave de los mismos previo a su anastomosis posterior o la interposición de un segmento de colon entre ellos. Las complicaciones agudas más frecuentes de esta intervención son la deshiscencia de la anastomosis y la formación de estenosis. Incluso tras una reparación correcta suele haber problemas de alimentación, debidos a la escasa motilidad del segmento esofágico distal o a un reflujo gastroesofágico. En estos casos, y si el tratamiento médico fracasa, puede ser necesaria una fundoplicatura de Nissen antes de introducir la alimentación oral.
Hernia diafragmática
Protrusión del contenido abdominal hacia el tórax a través de un defecto del diafragma.
La hernia diafragmática es mucho más frecuente en la parte posterolateral del diafragma (hernia del agujero de Bochdalek) y se sitúa en el lado izquierdo en el 90% de los casos. A través del defecto pueden introducirse en el hemitórax afectado las asas del intestino, incluso la mayor parte del contenido abdominal. Si la hernia es grande, el pulmón del lado afectado será hipoplásico. Después del nacimiento, cuando el RN llora y deglute aire, las asas intestinales se llenan de aire con rapidez y su dilatación puede provocar una dificultad respiratoria aún mayor, al empujar el corazón y las estructuras mediastínicas hacia la derecha, comprimiendo al pulmón derecho normal. En los casos graves, aparece sufrimiento respiratorio inmediatamente después del parto y con un estado de suma gravedad. Además del gran compromiso de la función respiratoria, el niño presentará un abdomen escafoideo (debido al desplazamiento de muchas de las vísceras abdominales hacia el tórax). En el hemitórax afecto, generalmente el izquierdo, pueden auscultarse ruidos intestinales (y ausencia de ruidos respiratorios). En los casos menos graves, aparece una dificultad espiratoria leve varias horas o días después, a medida que el contenido abdominal se hernia a través del defecto.
La combinación de un pulmón hipoplásico y otro atelectásico y el frecuente desarrollo de una hipertensión pulmonar primaria causa un aumento de la resistencia vascular pulmonar, con disminución del flujo sanguíneo pulmonar y aparición de un cortocircuito derecha-izquierda bien a través del foramen oval, bien por el conducto arterioso permeable, con una hipoxemia intensa.
Recientemente se comprobó que la hernia diafragmática congénita se asocia a hipoplasia y engrosamiento anómalo de las arteriolas pulmonares, con una gran resistencia al flujo a través de los vasos pulmonares (hipertensión pulmonar persistente) que impide una oxigenación adecuada. El riego sanguíneo pulmonar es tan escaso que el lactante se mantiene hipóxico, incluso aunque reciba respiración asistida. La hipertensión pulmonar persistente es la causa más importante de muerte en los niños con hernia diafragmática congénita.
Diagnóstico
Cuando es muy pequeño, el defecto diafragmático se identifica en las radiografías de tórax sólo como una pequeña porción de intestino que protruye en el tórax. Cuando es mayor, el hallazgo característico es la presencia de numerosas asas intestinales llenas de aire que ocupan el hemitórax, con desplazamiento contralateral del corazón y de las estructuras mediastínicas. Si la radiografía se obtiene inmediatamente después del parto, antes de que el niño haya deglutido aire, el contenido abdominal aparecerá como una masa opaca situada en el hemitórax.
Tratamiento
El RN deberá ser intubado y ventilado de inmediato en la sala de partos, pues la ventilación con bolsa y mascarilla agrava progresivamente el compromiso respiratorio, a medida que las vísceras se llenan de aire. La aspiración continua del estómago con una sonda nasogástrica de doble luz impide que el aire deglutido progrese hacia los tramos inferiores del intestino y contribuya a la compresión del pulmón. En caso necesario, la parálisis con norcuron o pancuronio puede facilitar la ventilación y evitar la deglución de aire. Es imprescindible una intervención quirúrgica que introduzca el intestino en el abdomen y cierre el defecto diafragmático.
La constricción de los vasos pulmonares puede contrarrestarse mediante alcalinización, administrando bicarbonato sódico i.v. La inhalación de óxido nítrico puede ayudar a dilatar las arterias pulmonares y a mejorar la oxigenación sistémica; este tratamiento es experimental. Algunos lactantes con hernia diafragmática e hipertensión pulmonar persistente que no reacciona a la respiración asistida y a la alcalinización pueden responder a la oxigenación con membrana extracorpórea (OMEC, v. Hipertensión pulmonar persistente del RN en el cap. 260), aunque los que tienen una hipoplasia pulmonar extrema no sobrevivirán. Más que operar de urgencia, es preciso aplicar todas las medidas médicas, incluyendo la inhalación de óxido nítrico, el uso de un ventilador de alta frecuencia y la OMEC, si es necesaria, para estabilizar correctamente al niño antes de la cirugía. El transporte de un RN inestable en estado muy grave con una hernia diafragmática congénita e hipertensión pulmonar persistente es muy difícil. Por tanto, cuando la hernia se diagnostica por ecografía antes del nacimiento, es aconsejable que el parto tenga lugar en un centro que disponga de OMEC y de un programa de cirugía pediátrica.
Estenosis hipertrófica del píloro
Obstrucción de la luz pilórica debida a la hipertrofia muscular del píloro.
La estenosis hipertrófica del píloro puede causar la obstrucción casi completa del tracto de salida del estómago. Rara vez presente en el momento del nacimiento, suele desarrollarse a lo largo de las 4-6 primeras semanas de vida, y los signosde obstrucción no suelen manifestarse hasta ese momento. Hacia el final del primer mes de vida, el lactante presenta vómitos alimenticios en proyectil, no teñidos de bilis. A veces, se observan a simple vista las ondas peristálticas cruzando el epigastrio de derecha a izquierda. El retraso del diagnóstico puede conducir a vómitos de repetición, deshidratación, fracaso del incremento ponderal y alcalosis metabólica hipoclorémica (causada por la pérdida de ácido clorhídrico).
El diagnóstico se establece por la palpación de una pequeña masa pilórica de 2 a 3 cm, firme y móvil, parecida a una aceituna, situada en la profundidad del lado derecho del epigastrio o por la identificación del músculo pilórico hipertrófico en una ecografía abdominal. En caso de duda, el tránsito digestivo con papilla de bario demostrará un vaciado gástrico lento y el típico «signo de la cuerda», debido a una luz pilórica muy larga y estrecha.
El tratamiento de elección es la piloromiotomía longitudinal, que deja intacta la mucosa y separa las fibras del músculo seccionado. El lactante suele comenzar a tolerar la alimentación oral algunos días más tarde.
Obstrucción duodenal
Entre sus posibles causas se encuentran la atresia, la estenosis y la compresión por una masa extrínseca. Después del íleon, el duodeno es la localización más frecuente de la atresia intestinal primaria (v. más adelante). Esta obstrucción es más frecuente en los niños con síndrome de Down. Los RN con anomalías de la rotación intestinal (v.más adelante) pueden presentar bandas peritoneales (bandas de Ladd) que cruzan el duodeno y producen su oclusión parcial o total; estos lactantes corren un gran riesgo de desarrollar un vólvulo del intestino medio, que es una urgencia quirúrgica (v. más adelante). También el quiste del colédoco y el páncreas anular pueden causar una obstrucción duodenal por compresión extrínseca. Los niños con quiste del colédoco tienden asimismo a presentar grados variables de ictericia obstructiva.
Síntomas, signos, diagnóstico y tratamiento
Como se trata de una obstrucción alta, suele existir el antecedente de hidramnios, y el RN presenta vómitos biliosos enérgicos después de las primeras tomas. Las radiografías simples muestran el típico signo de la doble burbuja: una burbuja grande, llena de aire, que corresponde al estómago, y una segunda burbuja, correspondiente al duodeno dilatado y lleno de aire próximo a la zona ocluida, a partir de la cual el aire es muy escaso o nulo. El tránsito intestinal con contraste permite identificar el lugar de la obstrucción y la presión extrínseca causada por un quiste del colédoco, mientras que el enema de bario revela la malrotación, en la que la obstrucción se debe a las bandas peritoneales (v. más adelante).
Ante la sospecha de una obstrucción duodenal, debe suspenderse la alimentación del lactante. La aspiración continua con una sonda nasogástrica de doble luz permite descomprimir el estómago y previene los vómitos y la consiguiente aspiración. Después de efectuar las pruebas diagnósticas adecuadas debe practicarse la cirugía para corregir la obstrucción.
OBSTRUCCIÓN DEL INTESTINO DELGADO DISTAL Y DEL INTESTINO GRUESO
En casi todos los casos no existen antecedentes de hidramnios materno, ya que gran parte del líquido amniótico deglutido fue absorbido por el intestino delgado proximal a la obstrucción. Las primeras tomas suelen tolerarse bien, pero al final del primer o segundo días comienza a desarrollarse una distensión abdominal que se asocia a vómitos biliosos o fecaloideos. El lactante puede defecar una pequeña cantidad inicial de meconio, pero luego ya no expulsa más heces.
Puede ser útil examinar el meconio con un microscopio para buscar la presencia de células escamosas y lanugo (prueba de Farber), normalmente presentes porque también lo están en el liquido amniótico; su hallazgo hace que la obstrucción intestinal completa sea menos probable, pero no descarta del todo la atresia intestinal, ya que un intestino previamente permeable puede obstruirse en fases posteriores de la gestación (p.ej., por vólvulo). El enfoque diagnóstico general y el tratamiento preoperatorio consisten en interrumpir la alimentación oral, introducir una sonda nasogástrica de doble luz para impedir la distensión progresiva del aparato GI o la posible aspiración de un vómito, corregir las alteraciones hidroelectrolíticas, obtener una radiografía simple y, a continuación, realizar un enema opaco para delimitar la anatomía.
Síndrome del tapón meconial
Obstrucción intestinal del lactante debida a un meconio espeso que forma un molde en la luz del colon.
Aunque este síndrome suele producirse en niños por lo demás normales, es más frecuente en los hijos de madres diabéticas o de madres toxémicas tratadas con sulfato de magnesio. El meconio espeso, condensado y gomoso forma un molde del colon y puede causar una obstrucción completa, con imposibilidad de expulsar las heces, distensión y vómitos. El enema con meglumina diatrizoato dibuja los bordes del molde y suele faciilitar su separación y eliminación. Aunque la mayor parte de los lactantes son normales, puede ser convenientellevar a cabo estudios diagnósticos para identificar una posible enfermedad de Hirschsprung (v.más adelante) o una fibrosis quística (v. también cap. 267), si existen otros hallazgos sugestivos o una historia familiar compatible.
Íleo meconial
Obstrucción intestinal debida a la presencia de un meconio sumamente pegajoso que se adhiere a la mucosa del íleon terminal.
El íleo meconial es casi siempre una manifestación precoz de la fibrosis quística (v. también cap. 267). El meconio adherente del íleo meconial se distingue fácilmente del tapón meconial elástico descrito más arriba; se adhiere a la mucosa intestinal y causa la obstrucción del íleon terminal. Distal a la obstrucción, el colon es estrecho y contiene bolitas de meconio seco. Este colon relativamente vacío y de pequeño diámetro recibe el nombre de «microcolon». A veces, las asas intestinales pueden palparse a través de la pared abdominal y muestran una consistencia pastosa característica.
Síntomas, signos y diagnóstico
Las asas del intestino delgado, distendidas por el meconio espeso y adherente, pueden girar alrededor de sí mismas y dar lugar a un vólvulo intrauterino. Si el intestino pierde su riego vascular y sufre un infarto, se producirá una peritonitis meconial estéril, que a menudo puede sospecharse por la presencia de restos de meconio calcificado cubriendo las superficies peritoneales e incluso el escroto en las radiografías. El asa intestinal necrosada puede reabsorberse, dejando una o más áreas de atresia intestinal, o puede ser aislada del resto para formar un gran quiste lleno de líquido.
El diagnóstico se confirma por la presencia de proteína digerida en el meconio (agitar y centrifugar una mezcla meconio: agua [1:1] y añadir ácido tricloroacético al 10% al sobrenadante. La aparición de un precipitado blanco abundante indica la presencia de albúmina no digerida). También existen tiras reactivas comercializadas para investigar la presencia de albúmina en el meconio. El contenido intraluminal puede aparecer granuloso en las radiografías simples, debido a las pequeñas burbujas de aire que están mezcladas con el meconio. Una prueba del sudor positiva confirmará el diagnóstico de fibrosis quística. Puede emplearse una sonda HISF (hibridación in situ con fluorescencia) para identificar una de las mutaciones genéticas que suelen asociarse a la enfermedad; sin embargo, no todas las mutaciones son identificables, por lo que sólo la negatividad de la prueba del sudor permite descartar definitivamente la mucoviscidosis.
Pronóstico y tratamiento
Los niños que sobreviven a un episodio de íleo meconial secundario a una fibrosis quística no sufren enfermedades pulmonares más graves que los demás pacientes con la enfermedad. Ante un diagnóstico de certeza o sospecha de íleo meconial en un caso no complicado (p. ej., sin perforación, vólvulo o atresia), la obstrucción puede resolverse administrando uno o varios enemas con un medio de contraste diluido (p. ej., una solución diluida de meglumina diatrizoato y diatrizoato sódico), bajo control radioscópico. Las grandespérdidas GI de agua secundarias a la presencia de un medio de contraste hipertónico deben ser corregidas i.v. para prevenir una deshidratación rápida con shock. Para disolver y eliminar el meconio anormal y corregir la obstrucción suele ser necesario hacer una ileostomía en cañón de escopeta y el lavado repetido con N-acetilcisteína de las asas proximales y distales.
Atresia intestinal
La atresia intestinal es más frecuente en el íleon, seguida del duodeno, el yeyuno y el colon (las obstrucciones duodenal y yeyunal se trataron más arriba). La atresia ileal suele manifestarse al final del primer día de vida o durante el segundo. El cuadro consiste en distensión abdominal progresiva, ausencia de defecación y, por último, regurgitación del alimento. El enfoque diagnóstico general y el tratamiento preoperatorio son similares a los de otros tipos de obstrucción del intestino delgado distal o del intestino grueso (v. más arriba). El pronóstico es bueno.
Durante la intervención quirúrgica debe examinarse la totalidad del intestino, para descartar la presencia de áreas múltiples de atresia; la porción atrésica se extirpa y, en general, se hace una anastomosis terminoterminal. Cuando ello es imposible porque el extremo proximal está muy dilatado y el distal, no utilizado, es muy estrecho, resulta a veces más seguro efectuar una ileostomía en cañón de escopeta y retrasar la anastomosis hasta que el diámetro del intestino proximal disminuya. Existen distintas técnicas quirúrgicas para anastomosar dos asas intestinales de distinto tamaño.
Anomalías de la posición del intestino secundarias a malrotación
Durante el desarrollo embrionario, el intestino primitivo sale fuera de la cavidad abdominal. Cuando vuelve a ella, el intestino grueso suele rotar en sentido contrario a las agujas del reloj hasta que el ciego alcanza la fosa ilíaca derecha. La rotación incompleta, en la que el ciego ocupa cualquier otra posición (por lo general, el hipocondrio derecho o el epigastrio), puede causar una obstrucción intestinal debida a las bandas retroperitoneales que cruzan el duodeno o a un vólvulo del intestino delgado, pues éste, al carecer de su fijación peritoneal normal, gira sobre su mesenterio, estrecho y largo. La presentación clínica inicial es similar a la de cualquier otra obstrucción intestinal. El niño comienza a vomitar bilis y se encuentra gravemente enfermo. Si las radiografías simples de abdomen revelan una escasa cantidad de gas intestinal distal al duodeno y sugieren un vólvulo del intestino medio, deberá procederse a un diagnóstico y tratamiento de urgencia. Con el enema de bario se comprueba que el ciego no se halla en la fosa ilíaca derecha. El tránsito intestinal típico de un paciente con vólvulo demuestra la torsión del intestino (es decir, el lugar de la obstrucción) al nivel del ligamento de Treitz. El vólvulo es una urgencia quirúrgica porque el asa intestinal afectada se gangrenará si la obstrucción no se resuelve con rapidez.
Enfermedad de Hirschsprung
(Megacolon congénito)
Alteración congénita de la inervación de los segmentos inferiores del intestino, generalmente limitada al colon, que produce una obstrucción funcional total o parcial.
La enfermedad de Hirschsprung se debe a la ausencia congénita de los plexos autónomos de Meissner y Auerbach de la pared intestinal, habitualmente limitada al colon distal. La actividadperistáltica del segmento afectado es nula o anormal, produciendo un espasmo continuo del músculo liso y una obstrucción parcial o completa, con acumulación del contenido intestinal y dilatación masiva del intestino proximal, que posee una inervación normal. Aunque su localización más frecuente es la anal, la obstrucción puede extenderse proximalmente y afectar a segmentos variables del colon, a la totalidad del mismo y, muy raras veces, al íleon terminal o a todo el aparato GI. Las lesiones discontinuas, «a saltos», son muy raras. El lactante presenta ausencia de deposiciones, distensión abdominal y, por último, vómitos. En ocasiones, en el lactante con aganglionosis limitada al ano, se encuentra tan sólo un estreñimiento leve o intermitente, a menudo con episodios intercurrentes de diarrea leve, en cuyo caso es posible que la enfermedad no se diagnostique hasta una etapa posterior de la lactancia. No obstante, es importante establecer el diagnóstico correcto lo antes posible. Cuanto más tiempo permanezca la enfermedad sin tratar, mayor será el riesgo de desarrollar una enterocolitis tóxica (megacolon tóxico), que puede ser fulminante y mortal. Casi todos los casos se diagnostican durante los primeros días de la lactancia. En los lactantes de mayor edad, los síntomas y signos comprenden anorexia, ausencia de la necesidad fisiológica de defecar, un recto vacío en la exploración, un colon palpable y una peristalsis visible. También puede haber retraso del crecimiento.
Diagnóstico y tratamiento
El enema opaco revela una transición entre el diámetro del colon proximal dilatado proximal a la obstrucción y el segmento distal, estrecho, que carece de inervación normal. Sin embargo, en el período neonatal pueden no encontrarse los signos característicos. La biopsia rectal permite el diagnóstico definitivo, pues demuestra la ausencia de ganglios nerviosos y el aumento del número de fibras que se tiñen con la colinesterasa.
La enfermedad de Hirschsprung neonatal debe tratarse mediante un colostomía en un lugar del colon proximal al segmento agangliónico. La extirpación de toda la porción anormal del colon y la corrección definitiva con una intervención de descenso pueden aplazarse hasta que el paciente sea mayor y el colon y el ano más grandes. No obstante, algunos cirujanos llevan a cabo uno de los distintos procedimientos definitivos existentes ya en el período neonatal. Una vez corregido el defecto, el pronóstico es bueno; casi todos los niños desarrollan un control satisfactorio delos movimientos intestinales.
Enterocolitis tóxica
(Megacolon tóxico)
La enterocolitis tóxica puede deberse a la ausencia de deposiciones de la enfermedad de Hirschsprung, cuando la proliferación de las bacterias en el segmento distendido trae consigo la formación de abundantes toxinas bacterianas. El consiguiente paso masivo y fulminante de líquido hacia la luz intestinal o en forma de diarrea produce una pérdida extrema de líquidos y electrolitos y puede causar la deshidratación y la muerte con gran rapidez. La reposición de líquidos y los antibióticos son importantes, pero pueden no prevenir la muerte, a menos que la obstrucción se resuelva con rapidez mediante una colostomía. Los lavados salinos del colon por medio de una sonda rectal pueden ser útiles en las fases iniciales de estabilización, antes de proceder a la cirugía.
Atresia anal
La atresia anal es evidente en la exploración física habitual, ya que el ano no es permeable. Si el diagnóstico pasara inadvertido en el examen de rutina del RN y éste fuese alimentado, aparecerían con rapidez todos los signos de obstrucción del intestino distal. Los varones con atresia anal presentan a menudo una fístula desde la bolsa anal hasta la uretra o el periné. En las niñas, la fístula se extiende desde la bolsa anal hasta la vagina, el frenillo o, más rara vez, la vejiga. El ano ciego y la piel del periné pueden hallarse a varios cm de distancia o estar separados sólo por una delgada membrana de piel que cubre el orificio anal. Debe filtrarse y examinarse la orina para buscar la presencia de meconio o bacterias, que indicarían una comunicación con el aparato urinario. Las radiografías simples y las «fistulografías» con el paciente en decúbito prono lateral permiten establecer el nivel de la lesión. La presencia de una fístula cutánea suele indicar una atresia baja, en cuyo caso puede ser posible la corrección definitiva con un abordaje perineal. Sin fístula perianal es más probable una lesión alta, que casi con seguridad precisaráuna colostomía, aplazándose la corrección definitiva hasta que el lactante sea mayor y las estructuras a reparar más grandes. Hasta entonces se hará una colostomía para resolver la obstrucción.
DEFECTOS DEL CIERRE DE LA PARED ABDOMINAL
Onfalocele
Protrusión de cantidades variables de vísceras abdominales a través de un defecto de la línea media localizado en la base del ombligo.
En el onfalocele, la herniación está cubierta por una fina membrana y puede ser pequeña (algunas asas intestinales) o contener la mayor parte de las vísceras abdominales (intestino, estómago e hígado). Los riesgos inmediatos son la desecación de las vísceras, la hipotermia debida a la pérdida decalor y la deshidratación por evaporación del agua a partir de las vísceras expuestas y la infección de las superficies peritoneales. Los RN con onfalocele presentan una incidencia de otras anomalías superior a la habitual, incluyendo las atresias intestinales y las malformaciones congénitas y renales, que deben ser identificadas y valoradas antes de proceder a la corrección quirúrgica.
Inmediatamente después del parto, la zona herniada debe cubrirse con esponjas empapadas con suero fisiológico, cubiertas a su vez con un vendaje oclusivo para mantener la esterilidad y evitar la evaporación, lo que también permitirá evitar la hipotermia por pérdida de calor. Otra técnica para evitar la evaporación consiste en colocar al RN en una bolsa intestinal estéril, que contiene suero fisiológico estéril y tibio.
Después se destinará el tiempo necesario a la evaluación del niño, para buscar otras anomalías asociadas, sobre todo las que conllevan mayor riesgo de mortalidad, antes de proceder a la cirugía. El cierre primario se lleva a cabo lo antes posible. En los onfaloceles grandes, la cavidad abdominal puede ser demasiado pequeña para contener todos los órganos. En estos casos, las vísceras se cubren con una bolsa o chimenea hecha con silicona polimerizada (Silastic), cuyo tamaño se va reduciendo poco a poco a lo largo de varios días, a medida que la capacidad abdominal aumenta lentamente, hasta que todas las vísceras se mantienen en el interior de la cavidad.
Gastrosquisis
Protrusión de las vísceras abdominales a través de un defecto de la pared abdominal, habitualmente situado a la derecha del ombligo.
En la gastrosquisis, las vísceras herniadas no están cubiertas por una membrana y el intestino se encuentra muy edematoso y eritematoso, a menudo cubierto por una red de fibrina. Estos hallazgos indican una inflamación duradera, debida a la larga exposición directa del intestino al líquido amniótico (peritonitis química). Los RN con gastrosquisis no tienen mayor incidencia de otras malformaciones, excepto la inevitable malrotación intestinal y, posiblemente, otras anomalías intestinales, incluyendo la atresia. No suele ser necesario hacer una evaluación detallada; estos pacientes pueden ser tratados quirúrgicamente una vez estabilizados. La cirugía es similar a la del onfalocele. Suelen transcurrir varias semanas hasta que el intestino recupere su función y pueda iniciarse la alimentación oral; algunos lactantes tienen problemas muy prolongados debidos a las alteraciones de la movilidad del intestino.
URGENCIAS QUIRÚRGICAS DIVERSAS
Las hernias inguinales son más frecuentes en los varones, sobre todo en los prematuros. Dada su tendencia a la incarceración, las hernias inguinales deben corregirse precozmente, en general cuando el niño pesa ³2,2 kg. Por el contrario, las hernias umbilicales no suelen incarcerarse, se cierran espontáneamente al cabo de varios años y no suelen ser susceptibles de reparación quirúrgica.
La invaginación (prolapso de un asa intestinal en el interior de otra), aunque rara en el RN, es una urgencia, debido al riesgo de gangrena del intestino. En ocasiones excepcionales se producen grandes infartos intestinales por oclusión de una arteria mesentérica, a causa de trombos murales o émbolos, tras la colocación alta de un catéter en la arteria umbilical.
La perforación gástrica del RN es a menudo espontánea y puede ser debida a un defecto congénito del estómago, en general a lo largo de la curvadura mayor. Se produce una distensión brusca del estómago y las radiografías abdominales muestran un neumoperitoneo masivo. Los lactantes prematuros tratados con corticosteroides por una atresia broncopulmonar corren mayor riesgo de ulceración péptica, con hemorragia GI alta o perforación gástrica o duodenal; el uso de un bloqueante H2 (p.ej., ranitidina) eleva el pH del estómago en estos niños e inhibe la producción de HCl, reduciendo el riesgo. El pronóstico suele ser bueno tras la corrección quirúrgica de la perforación.
Se han descrito perforaciones ileales en prematuros tratados con indometacina para cerrar un conducto arterioso persistente; parece probable que este cuadro se deba a una isquemia local secundaria a la vasoconstricción inducida por la indometacina.
ATRESIA BILIAR Y HEPATITIS NEONATAL
La atresia biliar se debe a la obstrucción del árbol biliar por esclerosis progresiva del colédoco. En casi todos los casos, se desarrolla varias semanas después del nacimiento, probablemente tras un proceso de inflamación y cicatrización fibrosa de los conductos extrahepáticos (y, a veces, también los intrahepáticos). Es muy rara en los mortinatos y en los RN. La causa de la respuesta inflamatoria se desconoce; ocasionalmente, se ha achacado la responsabilidad a un virus específico.
El síndrome de hepatitis neonatal (hepatitis de células gigantes) suele ser idiopático, aunque en raras ocasiones se han descrito casos asociados al citomegalovirus, al virus de la hepatitis B o a una deficiencia de a1-antitripsina.
Síntomas, signos y diagnóstico
Parece probable que la atresia biliar y la hepatitis neonatal formen parte de un mismo trastorno, más que ser entidades distintas. En ambas, la ictericia colestásica con hiperbilirrubinemia mixta, las orinas oscuras (bilirrubina conjugada), las heces acólicas y la hepatomegalia suelen aparecer unas 2 sem tras el nacimiento. A los 2 o 3 meses de edad, el niño suele presentar retraso del crecimiento, irritabilidad por prurito y signos de hipertensión portal.
Puede ser difícil distinguir la atresia biliar de la hepatitis neonatal. En ambas, las pruebas de función hepática reflejan la colestasis y la inflamación hepatocelular. Las pruebas diagnósticas adecuadas suelen permitir la exclusión de otras causas de ictericia obstructiva neonatal (p. ej., infecciones específicas, deficiencia de a1-antitripsina, galactosemia, mucoviscidosis). Las cifras de bilirrubina total y directa, AST, ALT y fosfatasa alcalina, junto a los niveles séricos de ácidos biliares, muchas veces no permiten diferenciar una atresia biliar de una hepatitis neonatal con colestasis intensa. La ausencia de vesícula biliar identificable y de conductos biliares extrahepáticos en la ecografía es muy sugestiva de atresia biliar; este procedimiento puede demostrar también que un quiste del colédoco está comprimiendo y obstruyendo las vías biliares. La colocación de un sonda nasoduodenal para obtener un muestra de jugo duodenal recogida por gravedad puede revelar la presencia de bilis (también es posible medir la bilirrubina); la excreción de bilis contradice firmemente el diagnóstico de atresia biliar completa. La escintigrafía hepática con tecnecio 99m PIPIDA (N-para-isopropil-acetanilida-ácido iminodiacético) resulta útil para identificar el flujo biliar extrahepático. En caso de duda, la biopsia hepática percutánea es la prueba diagnóstica más exacta (v.cap. 37) para diferenciar la hepatitis neonatal de la atresia biliar, pero debe ser interpretada por un patólogo pediatra con experiencia.
Tratamiento
Ante la sospecha de atresia biliar, la laparotomía debe llevarse a cabo antes de los 2 meses de vida, pues, de lo contrario, se desarrollará una cirrosis biliar irreversible. En la intervencion se realiza una colangiografía intraoperatoria para delimitar el árbol biliar y se toma una biopsia hepática para su estudio por congelación. Sólo en el 5 a 10% de los niños puede lograrse una buena anastomosis de los conductos biliares atrésicos; en los restantes, una modificación del procedimiento de Kasai (hepatoportoenterostomía) consigue a menudo restablecer el flujo de la bilis. No obstante, muchos pacientes sufren problemas médicos crónicos importantes, tales como colestasis, colangitis ascendentes de repetición e hipocrecimiento, en muchos de ellos con mortalidad tardía. El trasplante hepático salva las vidas de algunos niños con insuficiencia hepática; la atresia biliar es la indicación pediátrica más frecuente para el trasplante. La colestasis secundaria a una hepatitis neonatal suele resolverse lentamente, pero puede existir una lesión hepática permanente, a la que ciertos niños no sobrevivirán. El tratamiento de sostén se expone en el capítulo 38.
ANOMALÍAS MUSCULOESQUELÉTICAS
A continuación se exponen algunos trastornos frecuentes e importantes del sistema musculo-esquelético del RN.
ANOMALÍAS CRANEOFACIALES
Los defectos del desarrollo del primer y segundo arcos viscerales, que forman los huesos faciales y los oídos durante el 2.º mes de la gestación, causan diversas anomalías craneofaciales. Estas malformaciones son el labio leporino y el paladar hendido, los síndromes de Treacher Collins (disostosis mandibulofacial), Goldenhar (displasia oculoauriculovertebral), Pierre Robin y Waardenburg, el hipertelorismo y las malformaciones del oído externo y medio. Los más frecuentes se exponen en la tabla 261-3. La mayor parte de los niños con anomalías craneofaciales tienen una inteligencia y desarrollo normales.

El labio leporino y el paladar hendido son los defectos más frecuentes del primer arco y se producen en 1 de cada 700 u 800 nacimientos. Entre las causas propuestas está el uso de benzodiacepinas durante las primeras etapas del embarazo. La hendidura puede afectar tan sólo al paladar blando o extenderse completamente a los paladares blando y duro, a la apófisis alveolar del maxilar y al labio. La forma más leve es la úvula bífida. El labio leporino aislado es, sobre todo, un problema estético.
El paladar hendido dificulta la alimentación y el desarrollo del lenguaje. El tratamiento precoz consiste en utilizar tetinas especiales para paladar hendido o dispositivos dentales que ocluyan el paladar y permitan la succión, y el uso de un biberón que permita dar la fórmula con una leve presión (p. ej., en frasco de plástico). El tratamiento definitivo es el cierre quirúrgico; sin embargo, la cirugía puede interferir con el desarrollo de los centros de crecimiento que rodean al premaxilar. La cirugía plástica mejora el trastorno, pero un tratamiento incorrecto dejará como secuelas voz nasal, un aspecto alterado por la deficiencia central de la cara y cierta tendencia a la regurgitación. Puede ser necesario el tratamiento dental, de ortodoncia, psiquiátrico y del lenguaje.
Defectos asociados a las mandíbulas pequeñas (síndromes de Pierre Robin y Treacher Collins). Estos defectos dificultan la alimentación y facilitan las crisis de cianosis, porque la lengua es posterior y puede obstruir la faringe. El primer problema puede resolverse mediante una sonda nasogástrica. Si la cianosis o los trastornos respiratorios persisten, pueden realizarse una traqueostomía o una intervención quirúrgica destinada a fijar la lengua en una posición adelantada. También está indicada la evaluación otológica, ya que estos síndromes pueden afectar también al oído.
DEFECTOS VERTEBRALES
La tortícolis congénita consiste en una desviación de la cabeza, visible ya al nacimiento. La causa más frecuente es la lesión traumática del cuello durante el parto (a veces incluso antes) que da lugar a hematoma, fibrosis y contracción del músculo esternocleidomastoideo (ECM). Otras causas son las alteraciones de la columna vertebral como el síndrome de Klippel-Feil (fusión de las vértebras cervicales) o la fusión del atlas al occipital (fusión atlanto-occipital). Los tumores del SNC, las parálisis bulbares y las alteraciones funcionales oculares son las causas neurológicas más importantes, pero que rara vez se manifiestan al nacimiento. (v. también Tortícolis espasmódica, cap. 59). Las fracturas, luxaciones o subluxaciones de la columna cervical (sobre todo C1 y C2) o las anomalías de la apófisis odontoides son causas raras pero graves, ya que pueden provocar una lesión nerviosa permanente. La tortícolis secundaria a un traumatismo obstétrico no se manifiesta al nacimiento, sino que aparece algunos días o semanas después, observándose una masa no dolorosa en el ECM, generalmente en su porción media. Los estudios de imagen de la región cervical ayudan a excluir las causas óseas, que son las que pueden precisar estabilización. Cuando la tortícolis se debe a un traumatismo obstétrico, el tratamiento indicado es la distensión pasiva del ECM (rotando la cabeza y flexionan del cuello lateralmente hacia el lado contrario).
Los defectos vertebrales son la rara escoliosis congénita y los más frecuentes defectos vertebrales aislados del tipo de hemivértebras, vértebras en «cuña» o en «mariposa». Deben sospecharse ante la presencia de anomalías cutáneas en la línea media posterior o de anomalías congénitas de las extremidades inferiores. Con el crecimiento, pueden causar graves deformidades, por lo que el tratamiento con férulas o corsés corporales debe iniciarse con rapidez. Si la incurvación progresa, habrá que recurrir a la cirugía. Son frecuentes las malformaciones renales asociadas, por lo que debe practicarse una ecografía o una PIV.
MALFORMACIONES DE LA CADERA, LA PIERNA Y EL PIE
(Para el metatarso aducido y el metatarso en varo, v. Trastornos del pie, cap. 270.)
La luxación congénita de la cadera es más frecuente en los lactantes de sexo femenino que en los de sexo masculino y en los que nacen por parto de nalgas. Parece ser secundaria a una laxitud de los ligamentos que rodean la articulación o a una mala posición dentro del útero. La luxación puede ser unilateral o bilateral. En el primer caso, la extremidad afectada es más corta y puede observarse una asimetría de los pliegues cutáneos del muslo. El signo principal de subluxación o de luxación es la incapacidad para separar por completo el muslo cuando la cadera y la rodilla permanecen en flexión. Esta anomalía se debe al espasmo de los abductores, a menudo presente incluso aunque la cadera no esté realmente luxada en el momento de la exploración. Si la cadera está luxada, la abducción y rotación externa del fémur producirán un chasquido audible o palpable cuando la cabeza femoral entra en el acetábulo (signo de Ortolani). Son más frecuentes los chasquidos más débiles. El problema puede resolverse en el plazo de uno o dos meses, pero es necesario vigilar al niño cuidadosamente. La luxación parcial o completa puede resultar difícil de detectar al nacimiento, por lo que se recomienda hacer, durante el primer año de vida, comprobaciones frecuentes de la limitación de la abducción de la cadera. Parece que la ecografía de la articulación es un método fiable para establecer el diagnóstico precoz. Sin embargo, la interpretación de las radiografías iniciales puede ser difícil y sólo sirven de ayuda cuando confirman la impresión clínica. Es esencial iniciar el tratamiento lo antes posible, ya que, en general, se puede reducir la cadera inmediatamente después del nacimiento de forma que el acetábulo adquiera una forma casi normal al crecer. Sin embargo, si el tratamiento se retrasa, las posibilidades de corrección sin cirugía se reducirán progresivamente. El tratamiento consiste en la aplicación de aparatos (férulas, arneses o pañales con abundantes compresas) que mantengan las caderas afectadas en abducción y rotación externa, lo que favorece que el acetábulo vaya adoptando su forma normal con el crecimiento.
La torsión femoral, ya sea interna (anteversión, con las rodillas dirigidas hacia dentro) o externa (retroversión, con las rodillas dirigidas hacia fuera), es típica de los RN, en algunos de los cuales puede ser llamativa. Cuando el lactante comienza a ponerse de pie y a caminar, la alteración se corrige espontáneamente, incluso en los casos más extremos. La posición en decúbito prono durante el sueño puede prolongar la retroversión. Debe considerarse la conveniencia de hacer un estudio radiológico o ecográfico para descartar una luxación de la cadera. El mantenimiento de una postura adecuada y la realización de ejercicios pasivos ayudan a mejorar la situación.
La luxación anterior de la rodilla con hiperextensión al nacimiento es un cuadro raro que requiere un tratamiento inmediato. Puede deberse a un desequilibrio hormonal (si existen mielodisplasia o artrogriposis) o a una postura intrauterina anómala. Suele asociarse a luxación homolateral de la cadera. Si, por lo demás, el lactante es normal, el tratamiento inmediato y diario con movimientos pasivos de flexión y una férula en flexión suele conseguir una rodilla funcional.
La incurvación y la torsión de la tibia son frecuentes al nacimiento y rara vez pueden considerarse patológicas. La excepción es la incurvación con alteraciones radiológicas consistentes en un canal medular esclerótico y estrecho (enfermedad de Blount), que se asocia a un gran riesgo de fracturas y seudoartrosis, por lo que, en este caso, se colocará una ortesis de protección.
En el pie equinovaro, la forma más frecuente de pie zambo, el pie se encuentra en flexión plantar, inversión y marcada aducción. Las deformidades de una mala posición intrauterina pueden simular un pie zambo, pero se corrigen pasivamente, cosa que no ocurre con las alteraciones patológicas. El mejor tratamiento es el ortopédico, comenzando ya al nacimiento con la aplicación de escayolas repetidas para normalizar la posición del pie. En los casos graves, y si las escayolas no son suficientes, debe recurrirse a la cirugía.
En el pie calcaneovalgo, el pie es plano o convexo, se encuentra en flexión dorsal y puede aproximarse fácilmente a la parte inferior de la tibia. El tratamiento precoz con escayola para colocar el pie en posición de equinovaro o el uso de un zapato corrector suele dar buenos resultados.
OTRAS ALTERACIONES DEL HUESO Y DEL CARTÍLAGO
(Las osteocondrodisplasias, las osteopetrosis y las osteocondrosis se estudian en el cap. 270.)
Las condrodistrofias (v. en Osteocondrodisplasias, cap. 270) son enfermedades que alteran la transformación del cartílago en hueso. La mejor conocida es la acondroplasia. Todas se caracterizan por enanismo (generalmente con un tronco de tamaño normal y extremidades cortas) y suelen asociarse a otras anomalías. El desarrollo mental suele ser normal. En la mayoría de las condrodisplasias es necesario un estudio radiológico de los huesos largos para poder hacer un diagnóstico exacto. Hay que descartar el hipotiroidismo. El tratamiento es de sostén.
Las mucopolisacaridosis (v. en Trastornos hereditarios del tejido conjuntivo, cap. 270) son similares a las condrodisplasias, pero en algunos tipos existe también afectación visceral y del SNC, con retraso mental. A menudo se observan acortamiento del tronco y contracturas de las extremidades. Las alteraciones óseas no son evidentes en los RN, sino que se van desarrollando a medida que el niño crece.
La osteogénesis imperfecta es una enfermedad que cursa con fragilidad ósea y que se debe a un defecto en la producción del colágeno que ocasiona fracturas repetidas por traumatismos mínimos. La gravedad varía considerablemente; el tipo neonatal (congénito) es el más grave. Las fracturas pueden darse intraútero y durante la fase del parto. El traumatismo durante la fase del parto puede provocar hemorragia intracraneal y el parto de un feto muerto. Al nacimiento, el cráneo es blando y parece un «saco de huesos». Los lactantes que nacen vivos pueden morir de manera súbita durante los primeros días o semanas. Los supervivientes desarrollan extremidades cortas y otras deformidades óseas. El desarrollo mental es normal, salvo que se haya dado lesión del SNC. La esclerótica es delgada, transparente y parece de color azul. Puede darse pérdida de la audición debida a la otoesclerosis. El tratamiento ortopédico, la fisioterapia y la terapia ocupacional se utilizan para la prevención de las fracturas y para mejorar la función. El tratamiento con difosfonato (pamidronato) administrado i.v. ha evidenciado un aumento de la densidad mineral ósea y una mejoría de la función motriz, y puede reducir el dolor óseo y la resorción ósea.
La hipofosfatasia congénita se debe a la ausencia de fosfatasa alcalina en el suero y se traduce en una falta difusa de depósitos de calcio en los huesos. Suele manifestarse con vómitos, escasa ganancia de peso y aumento del tamaño de las epífisis similares al observado en el raquitismo. Los pacientes que sobreviven a la lactancia presentan deformidades óseas y enanismo, aunque su desarrollo mental es normal. No existe ningún tratamiento eficaz.
AMPUTACIONES CONGÉNITAS
Deficiencias transversales o longitudinales de los miembros debidas a una inhibición primaria del crecimiento intrauterino o a una destrucción intrauterina secundaria de los tejidos embrionarios normales.
A menudo no se conoce la etiología, pero dos de las causas conocidas son los agentes teratógenos (p. ej., talidomida) y las bridas amnióticas. En las deficiencias transversales faltan todos los elementos distales a un nivel determinado y el miembro parece un muñón de amputación. En las deficiencias longitudinales se produce un maldesarrollo específico, por ejemplo, una ausencia parcial o completa del radio, del peroné o de la tibia. Los RN con deficiencias transversales o longitudinales de las extremidades pueden tener también huesos bífidos o hipoplásicos, sinostosis, duplicaciones, luxaciones y otros defectos óseos. La afectación puede abarcar a más de un miembro y los defectos pueden ser de distintos tipos en cada extremidad. Para determinar cuáles son los huesos afectados hay que recurrir a su estudio radiológico. Es raro que estas deficiencias se asocien a anomalías del SNC.
El tratamiento ha de adaptarse a cada caso concreto y consiste fundamentalmente en prótesis, más útiles en las deficiencias de las extremidades inferiores o cuando la ausencia de la extremidad superior es completa o casi completa. Si existe una cierta actividad en un brazo o una mano, sea cual sea la importancia de la malformación, antes de recomendar una prótesis o una intervención habrá de hacerse una cuidadosa valoración funcional. La amputación terapéutica de una porción o de la totalidad de un miembro sólo debe recomendarse una vez efectuada la valoración detallada de las implicaciones funcionales y psicológicas de la pérdida y cuando sea esencial para la adaptación de una prótesis.
Las prótesis de la extremidad superior deben ser diseñadas para que cubran el mayor número de necesidades posible, de forma que pueda reducirse al mínimo el número de aparatos protésicos adicionales. Los niños utilizan mejor las prótesis cuando se han adaptado a ellas a edades tempranas y han llegado a ser una parte integral de su propio cuerpo durante los años de desarrollo. Con un tratamiento ortopédico eficaz y el apoyo auxiliar adecuado, la mayoría de los niños con amputaciones congénitas pueden llevar una vida normal.
ARTROGRIPOSIS MÚLTIPLE CONGÉNITA
(Contracturas congénitas múltiples)
Síndrome complejo caracterizado por múltiples contracciones articulares (sobre todo de las extremidades superiores y del cuello) sin otras malformaciones congénitas graves y con una inteligencia relativamente normal.
La artrogriposis consiste en la anquilosis prenatal de las articulaciones en flexión (contracción). Cuando el niño presenta una anquilosis generalizada al nacer, se habla de artrogriposis múltiple congénita (AMC), aunque también pueden encontrarse anquilosis de articulaciones aisladas.
Fisiopatología y etiología
El desarrollo articular tiene lugar en el 2.º mes de la gestación, y los trastornos que alteran la movilidad intrauterina (malformaciones del útero, gestaciones múltiples, oligohidramnios) pueden provocar artrogriposis.
La AMC se debe a trastornos del desarrollo neurológico, miopáticos o del tejido conjuntivo. Como causas de la amioplasia asociada se han propuesto las miopatías congénitas, la enfermedad de las células del asta anterior y la miastenia grave materna. La AMC no es un trastorno genético, aunque en algunas enfermedades genéticas (trisomía 18, espina bífida) la incidencia de artrogriposis es mayor.
Síntomas y signos
Los hombros suelen estar en aducción y en rotación interna, los codos en extensión y las muñecas y dedos en flexión. Las caderas pueden estar luxadas y suelen encontrarse en ligera flexión. Las rodillas están extendidas y los pies suelen adoptar una posición en equinovaro. Los músculos de las piernas son hipoplásicos y las extremidades tienden a ser tubulares y sin las características habituales. A veces se producen membranas de tejido blando en las caras ventrales de las articulaciones flexionadas. La columna puede ser escoliótica. Salvo por el escaso grosor de los huesos largos, el esqueleto es radiológicamente normal. Las minusvalías físicas pueden ser importantes. En general, la inteligencia es normal o muestra sólo una afectación ligera.
Otras malformaciones que pueden asociarse a la artrogriposis son microcefalia, paladar hendido, criptorquidia y malformaciones cardíacas y de la vía urinaria.
Diagnóstico, pronóstico y tratamiento
Es necesario descartar otras posibles malformaciones mediante un estudio completo. La electromiografía y la biopsia muscular ayudan al diagnóstico de los trastornos neuropáticos y miopáticos. La biopsia muscular muestra la amioplasia con sustitución del músculo por tejido fibroso o adiposo.
Las deformidades alcanzan su grado máximo al nacimiento, ya que la AMC no es progresiva y muchos niños evolucionan notablemente bien, de forma que dos terceras partes de los afectados llegan a caminar.
En todo caso está indicada una valoración ortopédica y fisioterapéutica precoz. Las manipulaciones articulares durante los primeros meses de vida pueden lograr importantes mejorías, a lo que también contribuyen las ortesis. En etapas posteriores puede ser necesario recurrir a la cirugía para alinear el ángulo de una anquilosis, aunque rara vez se logra aumentar la movilidad con ello.
DEFECTOS MUSCULARES
Un niño puede presentar ausencia congénita de un músculo o de un grupo muscular. La agenesia parcial o completa del pectoral mayor es uno de los defectos más frecuentes dentro de este grupo y puede observarse aislada o asociada a malformaciones de la mano del mismo lado (anomalía de Poland).
También puede apreciarse, ya al nacimiento, la ausencia de una o varias capas de la musculatura abdominal (síndrome del vientre en ciruela pasa, a menudo asociado a graves malformaciones genitourinarias, sobe todo hidronefrosis). Su incidencia es mayor en los varones, quienes a menudo presentan también criptorquidia. El pronóstico es reservado, incluso aunque elimine precozmente la obstrucción urinaria. La agenesia de musculatura abdominal suele ir acompañada de malformaciones del pie y del recto.
DEFECTOS NEUROLÓGICOS
Algunas de las malformaciones congénitas más graves del sistema nervioso (como anencefalia, encefalocele, espina bífida) se desarrollan en los dos primeros meses de la gestación y representan defectos de la formación del tubo neural (disrafias). Otras (como la hidranencefalia y la porencefalia) aparecen más tarde y parecen secundarias a procesos destructivos del encéfalo ya formado. Algunos defectos son relativamente benignos (p. ej., el meningocele).
En la actualidad, es posible la detección de muchas malformaciones durante la vida intrauterina gracias a la amniocentesis y a la ecografía (v.también cap. 247). El consejo genético a los padres de un niño con una anomalía neurológica grave es importante, pues el riesgo de tener otro hijo con el mismo defecto es elevado. Estos padres también precisan ayuda y apoyo psicológico. A las mujeres que han tenido un feto o un RN con un defecto del tubo neural se les debe prescribirun suplemento de ácido fólico (4 mg/d) antes de la concepción y durante las primeras fases de la gestación, ya que de este modo puede aminorarse sustancialmente el riesgo de defectos del tubo neural en gestaciones posteriores.
ANOMALÍAS ENCEFÁLICAS
La anencefalia o ausencia de hemisferios cerebrales es incompatible con la vida. A veces, el cerebro ausente es sustituido por un tejido nervioso quístico malformado que puede hallarse expuesto o cubierto de piel. Partes variables del tronco cerebral y de la médula espinal pueden faltar o estar malformadas. Todo esfuerzo diagnóstico o terapéutico es inútil, y estos pacientes nacen muertos o mueren al cabo de pocos días.
Pueden aparecer malformaciones de los hemisferios cerebrales. Los hemisferios pueden ser grandes, pequeños o asimétricos; las circunvoluciones pueden faltar, ser inusitadamente grandes o múltiples y pequeñas; los cortes microscópicos del cerebro de aspecto normal pueden presentar una desorganización de la disposición laminar neuronal normal. La disminución del tamaño de la cabeza (microcefalia) suele asociarse a estos defectos y normalmente va acompañada de retraso mental y motor entre moderado y grave. El tratamiento consiste en medidas generales de sostén y anticonvulsivantes, si son necesarios para controlar las convulsiones.
El encefalocele, o protrusión del tejido nervioso y meninges a través de un defecto del cráneo, se asocia a un cierre incompleto de la bóveda craneal (cráneo bífido). El encefalocele suele aparecer en la línea media y protruye hacia los conductos nasales a través del occipucio, aunque puede presentarse de forma asimétrica en las regiones frontal o parietal. Los encefaloceles pequeños pueden parecer cefalohematomas, pero la radiología demuestra la existencia de un defecto óseo en su base. La mayoría de los encefaloceles deben corregirse, ya que incluso los más grandes contienen fundamentalmente tejido nervioso heterotópico, que puede eliminarse sin provocar una incapacidad funcional importante. Cuando coexisten otras malformaciones graves, la decisión de corregir puede ser más difícil. La hidrocefalia (v.más adelante), a menudo asociada a un encefalocele, requiere un estudio con TC o ecografía y, si es progresiva, un tratamiento quirúrgico de derivación. Alrededor de la mitad de los RN afectados tienen otros defectos congénitos. En muchos de estos pacientes el pronóstico es bueno.
La porencefalia, un quiste o cavidad en el hemisferio cerebral que comunica con un ventrículo, puede formarse antes o después del nacimiento. El defecto se debe a una anomalía del desarrollo, una enfermedad inflamatoria o un accidente vascular como una hemorragia intraventricular que se extendió al parénquima. El examen neurológico suele ser anormal. El diagnóstico se confirma con TC o ecografía. La hidrocefalia progresiva puede precisar una intervención de derivación. El pronóstico es variable; algunos pacientes sólo desarrollan signos neurológicos menores y presentan una inteligencia normal.
La hidranencefalia es una forma extrema de porencefalia, en la que los hemisferios cerebrales se hallan casi totalmente ausentes. En general, el cerebelo y el tronco cerebral están bien formados y los núcleos de la base están intactos. Las meninges, los huesos y la piel que cubren la bóveda craneal son normales. Los resultados del examen neurológico neonatal suelen ser anormales, pero los hallazgos son sutiles. El desarrollo del niño es anormal. Externamente, la cabeza tiene un aspecto normal, pero con la transiluminación se observa que la luz brilla intensamente a través de ella. La TC o la ecografía confirman el diagnóstico. El tratamiento es de sostén, con derivación del LCR si el crecimiento del cráneo es excesivo.
La hidrocefalia, agrandamiento ventricular con acumulación excesiva de LCR, es la causa más frecuente de cabezas anormalmente grandes en los RN. Se produce cuando la producción de LCR supera a la reabsorción; suele deberse a una obstrucción en el acueducto de Silvio, aunque laobstrucción también puede localizarse en la salida del IV ventrículo (agujeros de Luschka y Magendie) o en los espacios subaracnoideos alrededor del tronco cerebral o encima de los hemisferios. La hidrocefalia puede ser comunicante o no comunicante. La primera se produce cuando el LCR fluye libremente hacia el espacio subaracnoideo y puede deberse a una obstrucción en el espacio subaracnoideo, aunque lo más habitual es que sea secundaria a una producción excesiva de LCR (sin obstáculos al flujo) debida a una inflamación meníngea por infección o por la presencia de sangre en el espacio subaracnoideo. La hidrocefalia no comunicante indica el bloqueo de algún punto del sistema ventricular o entre éste y el espacio subaracnoideo. El estudio complementario consiste en radiografías, ecografía y TC o RM del cráneo. Los estudios de imagen pueden mostrar separación de las suturas, áreas de adelgazamiento de los huesos o calcificaciones intracraneales (a menudo asociadas a infecciones congénitas). Las radiografías simples pueden revelar un aspecto de «metal batido» de los huesos (frecuente en los lactantes con mielomeningocele e hidrocefalia) que indica un aumento prolongado de la presión intracraneal. La TC pondrá de manifiesto el tamaño de los ventrículos y, posiblemente, el lugar de la obstrucción. La ecografía permite definir el grado de dilatación ventricular, y los estudios seriados documentan la progresión de la hidrocefalia. Los estudios ecográficos son especialmente valiosos después de una hemorragia intraven-tricular, pues la dilatación de los ventrículos puede ser transitoria, en cuyo caso sólo requiere tratamiento médico (v. más adelante). Cuando se sospecha una infección congénita, están indicados los estudios serológicos para Toxoplasma gondii, virus de la rubéola, Treponema pallidum, herpesvirus y citomegalovirus. En caso de convulsiones, el EEG puede ser útil. La evaluación posterior puede hacerse con análisis del LCR.
El diagnóstico diferencial debe hacerse con laslesiones intracraneales ocupantes de espacio (p.ej., hematomas subdurales, quistes porencefálicos, tumores), que pueden identificarse con la TC. También puede observarse la presencia de un cerebro anormalmente grande y, en general, malfuncionante (megalencefalia).
El tratamiento depende de la etiología; la administración de acetazolamida y glicerina o las punciones lumbares (si la hidrocefalia es comunicante) para reducir la presión del LCR pueden resultar transitoriamente útiles. Sin embargo, la hidrocefalia progresiva, sobre todo cuando el perímetro cefálico aumenta con excesiva rapidez, requiere una intervención de derivación del LCR para reducir la presión. Antes de la intervención es importante comprobar que se trata de una hidrocefalia progresiva verdadera, ya que a veces no progresa. El tipo de derivación depende de la experiencia y de las preferencias del neurocirujano: en general se prefieren las derivaciones ventriculoperitoneales a las ventriculoauriculares, pues tienen menos complicaciones. Una vez colocada la derivación, debe controlase el progreso del lactante, prestando atención al perímetro cefálico occipitofrontal, al desarrollo y al mayor riesgo de infecciones relacionadas con la derivación. Las exploraciones con TC o ecografías (si la fontanela anterior permanece abierta) permiten controlar el tamaño de los ventrículos. Algunos niños dejan de precisar la derivación cuando crecen, pero es difícil determinar el momento en que esto ocurre, por lo que rara vez se retiran. La cirugía fetal para tratar la hidrocefalia congénita antes del nacimiento no ha logrado resultados satisfactorios.
A menudo, la hidrocefalia se asocia a quistes de Dandy-Walker o a la malformación de Arnold-Chiari, ambas anomalías importantes de la fosa posterior.
Los quistes de Dandy-Walker son malformaciones del desarrollo en las que el IV ventrículo es quístico; suelen asociarse a hidrocefalia. El diagnóstico puede confirmarse por ecografía o TC, observando el desplazamiento superior de la fosa del seno lateral en las radiografías simples o transiluminando la fosa posterior. En general, la hidrocefalia requiere una derivación.
La malformación de Arnold-Chiari es un defecto en la formación del tronco cerebral. Las formas más extremas consisten en una elongación de las amígdalas cerebelosas, que suelen protruir a través del agujero occipital, incurvación de los colículos en forma de pico y engrosamiento de la médula espinal cervical superior. El diagnóstico se hace mediante TC o RM. El bloqueo de los orificios de salida del IV ventrículo o la estenosis asociada del acueducto de Silvio pueden determinar la producción de una hidrocefalia que precise una intervención para eliminar la obstrucción. La malformación de Arnold-Chiari puede aparecer aislada, pero a menudo se asocia a espina bífida o a siringomielia (v. cap. 182).
ESPINA BÍFIDA
Cierre defectuoso de la columna vertebral.
La espina bífida es una de las malformaciones más graves del tubo neural compatible con una vida prolongada. Su gravedad varía desde el tipo oculto sin hallazgos hasta la columna vertebral completamente abierta (raquisquisis) con incapacidad neurológica grave y muerte. En la espinabífida quística, el saco que protruye puede contener meninges (meningocele), médula espinal (mielocele) o ambas (meningomielocele). La espina bífida es más frecuente en las región dorsal baja, lumbar y sacra y suele abarcar de 3 a 6segmentos vertebrales. En el mielomeningocele, el saco suele estar compuesto por meninge, con una placa neural central. Si no está bien cubierto con la piel, el saco puede romperse fácilmente, aumentando el riesgo de meningitis.
Cuando la médula espinal o las raíces nerviosas lumbosacras se hallan involucradas en la espina bífida, como es habitual, aparecen grados variables de parálisis por debajo del nivel afectado. Dado que esta parálisis se produce ya en el feto, pueden observarse problemas ortopédicos (como pie zambo, artrogriposis o luxación de cadera; v.Defectos musculoesqueléticos, antes) en el momento del nacimiento. La parálisis suele alterar las funciones de la vejiga urinaria y del recto y la alteración resultante del aparato genitourinario puede, con el tiempo, lesionar gravemente los riñones. La cifosis, a veces asociada a la espina bífida, puede obstaculizar el cierre quirúrgico e impedir el decúbito supino. A menudo se observa hidrocefalia, que puede estar relacionada con una estenosis del acueducto o con una malformación de Arnold-Chiari (v. antes). También pueden encontrarse otras malformaciones congénitas.
Los exámenes complementarios comienzan con radiografías de la columna vertebral, cráneo, caderas y, si se hallan malformadas, de las extremidades inferiores. Es esencial estudiar el aparato urinario mediante análisis de orina, urocultivo, determinación de la creatinina sérica y del BUN, PIV y ecografía. La realización de otras pruebas dependerá de los defectos asociados y consistirá en estudios intracraneales (TC o ecografía) y del LCR.
El diagnóstico prenatal de la espina bífida se hace por la elevación de los niveles de a-fetoproteína en el suero materno y en el líquido amniótico. La ecografía puede mostrar los defectos óseos vertebrales y masas en los tejidos blandos.
El pronóstico depende del número y de la gravedad de las anomalías; es más grave en los pacientes que presentan parálisis total por debajo de la lesión, cifosis, hidrocefalia, hidronefrosis precoz y defectos congénitos asociados. Sin embargo, con una asistencia adecuada muchos niños evolucionan bien. La pérdida de la función renal y las complicaciones de la derivación son las causas habituales de muerte en los pacientes mayores con espina bífida.
La administración de suplementos de ácido fólico es la medida preventiva más importante (v.Defectos neurológicos, antes). El tratamiento requiere el esfuerzo conjunto de especialistas en varias disciplinas. Son importantes las evaluaciones iniciales neuroquirúrgica, urológica, ortopédica y de asistencia social. En general, antes de la intervención debe hacerse una valoración cuidadosa del RN para proporcionar el asesoramiento adecuado a la familia. Es importante valorar el tipo, la localización y la extensión de la lesión, el estado general de salud y las deficiencias asociadas del RN, las capacidades, los deseos y los recursos de la familia y los recursos comunitarios, incluyendola asistencia continuada. Tras ello puede tomarse la decisión sobre el alcance del tratamiento.
Si el paciente pierde LCR por el defecto, la cobertura antibiótica o la intervención neuroquirúrgica urgente reducirán el riesgo de infección meníngea y ventricular. La hidrocefalia puede precisar una intervención de derivación. Debe controlarse estrechamente la función renal y tratar rápidamente las infecciones urinarias. La uropatía obstructiva, tanto al nivel de la salida de la vejiga como de la uretra, debe tratarse enérgicamente, en especial cuando existe infección. La asistencia ortopédica debe comenzar precozmente y, en caso de pie zambo, con aplicación de una escayola. Hay que descartar la posible luxación de las caderas. Otras indicaciones ortopédicas continuas son la escoliosis, las fracturas patológicas, el desarrollo de úlceras de presión y la debilidad y espasmos musculares, que pueden causar nuevas deformidades.
DEFECTOS OCULARES CONGÉNITOS
GLAUCOMA CONGÉNITO
(Glaucoma del lactante, buftalmos, hidroftalmos)
Anomalía poco frecuente debida a un defecto congénito del ángulo iridocorneal de la cámara anterior que obstruye el flujo de salida del humor acuoso, provocando un aumento crónico de la presión intraocular.
El trastorno afecta a lactantes y niños y suele ser bilateral. El globo ocular aumenta considerablemente de tamaño; la córnea, de gran diámetro, se adelgaza, a veces se hace borrosa y puede protruir (en los lactantes, hay que vigilar estrechamente el diámetro y la transparencia de la córnea); la pupila puede ser grande y fija y la cámara anterior, profunda. Si la enfermedad progresa, se producirá la lesión del nervio óptico, con la consiguiente ceguera. El aspecto clave del tratamiento es la intervención quirúrgica precoz (gonotomía, goniopunción, trabeculotomía, trabeculectomía).
CATARATA CONGÉNITA
Opacificación del cristalino presente al nacimiento.
(Para las cataratas juvenil y del adulto, v. cap. 97.)
La catarata congénita puede ser consecuencia de anomalías cromosómicas, enfermedades metabólicas (p. ej., galactosemia), infecciones intrauterinas (p. ej., rubéola) y otras enfermedades maternas durante el embarazo. Las cataratas pueden ser nucleares y pasar inadvertidas si no se hace un examen del fondo del ojo en el momento del nacimiento. Si las cataratas son lo suficientemente densas como para enmascarar la visión del disco óptico y de los vasos, debe consultarse a un oftalmólogo para valorar su posible efecto en la visión.
La extirpación de la catarata en los primeros meses de la vida puede permitir el desarrollo de una fijación adecuada de la retina y las respuestas visuales de la corteza. La extracción de la catarata es técnicamente sencilla y en algunos pacientes puede implantarse una lente intraocular. La corrección visual posterior con gafas, lentillas de contacto o epiqueratofaquia (sutura de una córnea humana, torneada con una lente de contacto, sobre la córnea del receptor), es difícil, pero necesaria para lograr una buena visión. Una vez extraída una catarata unilateral, la calidad de la imagen en el ojo operado es inferior a la del ojo normal. Como el ojo normal suele quedar favorecido, el encéfalo suprime la imagen de peor calidad, lo que se traduce en un desarrollo insuficiente del sistema visual del ojo operado (ambliopia). Por tanto, para que el desarrollo de la visión sea normal en el ojo operado, es necesario un tratamiento enérgico y meticuloso de la ambliopia. Por el contrario, en las extracciones bilaterales, en las que la calidad de la imagen es similar en ambos ojos, ninguno de ellos resulta favorecido y el sistema visual se desarrolla por igual en los dos.
DEFECTOS RENALES Y GENITOURINARIOS
Las anomalías anatómicas congénitas del aparato genitourinario (GU) son más frecuentes que las de cualquier otro sistema orgánico. Sus complicaciones (obstrucción urinaria, estasis) pueden causar alteraciones de la función renal, infección y formación de cálculos y discapacidad sexual o esterilidad. El tratamiento suele ser quirúrgico.
RIÑÓN
Anomalías de fusión. Las anomalías de fusión (en las que los riñones están unidos, pero los uréteres penetran por separado en la vejiga) aumentan el riego de obstrucción al nivel de la unión pieloureteral, del reflujo vesicoureteral, displasia renal multiquística y lesiones por traumatismos abdominales anteriores.
La anomalía de fusión más frecuente es el riñón en herradura, en el que el parénquima renal situado a un lado de la columna vertebral se une con el del lado opuesto por los polos correspondientes (en general el inferior), dando lugar a un istmo de parénquima renal o de tejido fibroso que cruza la línea media. Los uréteres siguen un trayecto medial y anterior sobre este istmo y su drenaje suele ser normal. La obstrucción, cuando existe, suele ser secundaria a la inserción alta del uréter en la pelvis renal y no a su compresión en el istmo, lo que permite efectuar una pieloplastia sin necesidad de extirparlo.
La segunda anomalía de fusión por orden de frecuencia es la ectopia renal fusionada cruzada. El parénquima renal (representante de ambos riñones) se encuentra a un solo lado de la columna vertebral. Uno de los uréteres cruza la línea media y penetra en la vejiga en el lado opuesto al que ocupa el riñón. Cuando existe obstrucción de la unión pieloureteral, el tratamiento de elección es la pieloplastia.
El riñón pélvico fusionado (riñón en «torta») es una anomalía de fusión mucho menos frecuente, en la que una sola masa renal pélvica tiene dos sistemas colectores y dos uréteres. En caso de obstrucción, está indicada la reconstrucción.
Ectopia renal (localización anormal del riñón). Esta anomalía se debe a que el riñón no asciende desde su origen en la pelvis verdadera o a que asciende demasiado (riñón torácico). La incidencia de obstrucción de la unión pieloureteral es mayor en este tipo de riñones y lo mismo sucede con el reflujo vesicoureteral y la displasia renal multiquística en los riñones que han ascendido de forma incompleta, aunque no sucede igual en los que ascienden más de lo normal. El tratamiento es quirúrgico en los casos en que esté indicado.
Malrotación. Esta anomalía, generalmente menor, del eje renal puede dar una imagen anormal en la radiografía del sistema colector. Debe diferenciarse de los efectos de la obstrucción verdadera de las tumoraciones renales.
Agenesia. La agenesia renal bilateral (ausencia de ambos riñones, síndrome de Potter) es mortal. Se asocia a oligohidramnios, hipoplasia pulmonar, orejas de implantación baja. La agenesia renal unilateral no es rara y suele ir acompañada de agenesia ureteral con ausencia del trígono y del orificio ureteral vesical del mismo lado. La hipertrofia compensadora del riñón restante hace que la función renal sea normal.
Anomalías por duplicación. Los sistemas colectores supernumerarios pueden ser unilaterales o bilaterales y pueden implicar a la pelvis renal y a los uréteres (pelvis renal accesoria, pelvis y uréter doble o triple), a los cálices o a los orificios ureterales. El riñón doble (o riñón duplex) consiste en una masa renal única con más de un sistema colector. En estos casos existe mayor riesgo de ectopia ureteral con o sin ureterocele; el tratamiento depende de la anatomía y de la función de cada uno de los segmentos de drenaje. Puede ser necesario recurrir a la cirugía para corregir la obstrucción o el reflujo vesicoureteral.
Displasias renales. Estas anomalías parenquimatosas, con la consiguiente alteración de la función renal, son consecuencia del desarrollo anormal de la vascularización renal, de los túbulos o colectores o del aparato de drenaje. Para hacer el diagnóstico puede ser necesario recurrir a la biopsia.
Displasia renal multiquística. Esta unidad renal no funcionante está formada por quistes no comunicantes separados por un tejido sólido compuesto por haces fibrosos, túbulos primitivos y focos de cartílago. En general, se asocia a atresia ureteral y conlleva cierto riesgo de desarrollo de tumores, infecciones e hipertensión; algunos autores defienden la extirpación de estos riñones mientras que otros optan por la observación.
Hipoplasia renal. El riñón hipodesarrollado suele asociarse a una arborización incompleta de las yemas ureterales. El riñón es pequeño, pero posee nefronas histológicamente normales. En los casos de hipoplasia segmentaria puede haber una hipertensión asociada, por lo que a menudo está indicada la extirpación quirúrgica.
Enfermedad poliquística autosómica recesiva. Esta enfermedad, aunque rara (1/10.000 nacimientos), es la más frecuente de las lesiones quísticas renales (afecta también al hígado) determinadas genéticamente y a menudo da lugar a insuficiencia renal en la infancia. La enfermedad poliquística renal autosómica dominante se estudia en el capítulo 230.
Cuando la enfermedad se manifiesta en la primera infancia, sus síntomas son principalmente renales, mientras que cuando se manifiesta en la adolescencia, lo hace con síntomas de tipo hepático. Es probable que estas diferencias obedezcan a variaciones fenotípicas del mismo trastorno genético.
Los RN gravemente afectados suelen presentar una hipoplasia pulmonar secundaria a los efectos intrauterinos del oligohidramnios asociado. En los casos menos graves, los RN presentan un abdomen prominente con riñones enormes, firmes, de superficie lisa y simétricos. El hígado, aumentado de tamaño, es anormal, con fibrosis periportal, proliferación de conductos biliares y algunos quistes; el resto del parénquima hepático es normal. Estos hallazgos anatomopatológicos son los responsables de la hipertensión portal perisinusoidal con función hepática conservada o mínimamente afectada. El mejor método diagnóstico que, si se efectúa a finales de embarazo puede dar un diagnóstico intrauterino de presunción, es la ecografía.
Los pacientes de 5 a 10 años desarrollan signos de hipertensión portal como varices esofágicas y gástricas e hiperesplenismo (leucopenia, trombocitopenia). Si la enfermedad se manifiesta en la adolescencia, la nefromegalia es menos importante. La insuficiencia renal puede ser leve o moderada. Los síntomas más importantes son los dependientes de la fibrosis hepática progresiva (hipertensión portal, varices esofágicas y gástricas, insuficiencia hepática, hiperesplenismo). El diagnóstico es difícil, sobre todo en ausencia de antecedentes familiares conocidos. La ecografía puede mostrar los quistes renales o hepáticos; muchas veces, el diagnóstico definitivo se consigue mediante la biopsia.
Muchos RN mueren en los primeros días o semanas de vida a causa de una insuficiencia pulmonar. La mayoría de los que sobreviven a los primeros años desarrollan una insuficiencia renal progresiva. En los que tienen una afectación renalmenor se produce una insuficiencia hepática progresiva. Las derivaciones portocava o esplenorrenal reducen la morbilidad pero no la mortalidad. La experiencia con el trasplante es limitada en estos pacientes. Si se lleva a cabo, es necesario controlar el hiperesplenismo para evitar la inmunosupresión, ya que, de lo contrario, aquél provocará una leucopenia, con el consiguiente aumentodel riesgo de infección sistémica. La diálisis (v.cap. 223) tiene las mismas indicaciones que en los demás niños con insuficiencia renal crónica.
URÉTER
Las anomalías congénitas de los uréteres se asocian con frecuencia a anomalías renales, pero pueden aparecer de manera independiente. Entre sus complicaciones se encuentran la obstrucción, la infección y, a veces, la formación de cálculos debido a la estasis urinaria, así como incontinencia urinaria si los uréteres evitan la vejiga y desembocan en la uretra, el perineo o la vagina (en la mujer).
Anomalías por duplicación. La duplicación parcial o completa de uno o ambos uréteres puede asociarse a duplicación de la pelvis renal del mismo lado. El uréter procedente del polo superior del riñón desemboca en una localización más caudal que el uréter del polo inferior. Puede haber ectopia o estenosis de uno o de ambos orificios (v. antes), reflujo vesicoureteral en el uréter inferior o en ambos o un ureterocele. Para corregir el reflujo vesicoureteral, la obstrucción o la incontinencia urinaria, puede ser necesaria la cirugía.
Uréter retrocavo. Una anomalía del desarrollo de la vena cava (vena cava preureteral) hace que la porción infrarrenal de la vena pase por delante del uréter. El uréter retrocavo del lado izquierdo sólo se observa en los casos de persistencia del sistema de la vena cardinal izquierda o en el situs inversus completo. Puede provocar una obstrucción ureteral que, si es importante, se corregirá seccionando el uréter y haciendo una anastomosis uretero-ureteral anterior a la vena cava o al vaso iliaco.
Meatos ectópicos. Los meatos desplazados de los uréteres únicos o duplicados pueden observarse en la pared lateral de la vejiga, distalmente a lo largo del trígono, en el cuello vesical, en la uretra femenina distal al esfínter (lo que da lugar a incontinencia urinaria), en el sistema genital (próstata y vesícula seminal en el varón, útero o vagina en la mujer) o sobre la superficie corporal externa. Los meatos ectópicos laterales suelen asociarse a reflujo vesicoureteral, mientras que los distales tienden a asociarse a obstrucción e incontinencia. En estas últimas circunstancias, está indicada la cirugía, que, a veces, es también necesaria para corregir el reflujo vesicoureteral.
Estenosis. La estrechez congénita puede aparecer en cualquier parte del uréter, aunque la forma más frecuente afecta a la unión pieloureteral, y la menos frecuente a la unión vesicoureteral (megauréter primario). A menudo, la estenosis mejora con el tiempo y con el crecimiento del paciente, pero, si se produce una dilatación progresiva o aparecen infecciones, puede ser necesario reimplantar el uréter. Cualquier tipo de obstrucción constituye también una indicación quirúrgica.
Ureterocele. La protrusión del extremo inferior del uréter en la vejiga puede producir una dilatación obstructiva progresiva del mismo, que conduce a la ureterectasia, hidronefrosis, formación de cálculos, infección y pérdida potencial de la función renal. El tratamiento consiste en la incisión transureteral endoscópica o en una reparación abierta. Cuando el ureterocele afecta al polo superior de uréteres dobles, el tratamiento dependerá de la función de dicho segmento renal, ya que la incidencia de displasia renal es significativa en estos casos. Si no se detecta función renal o si se sospecha una displasia renal importante, puede ser preferible extirpar el segmento renal y el uréter afectados a reparar la obstrucción.
Reflujo vesicoureteral. El reflujo de la orina desde la vejiga hacia el uréter puede provocar una lesión de la vía urinaria superior por infección bacteriana y, a veces, por aumento de la presión hidrostática. Lo más frecuente es que su causa sea una alteración congénita del desarrollo de la unión vesicoureteral. El desarrollo incompleto del túnel ureteral intramural hace que la función de válvula del colgajo en la unión vesicoureteral sea insuficiente, permitiendo el retroceso de la orina vesical hacia el uréter y la pelvis renal. El reflujo puede existir incluso aunque el túnel sea suficiente cuando existe una obstrucción a la salida de la vejiga, con el consiguiente aumento de la presión intravesical o una disfunción vesical neurógena.
El reflujo transporta fácilmente las bacterias presentes en la vía urinaria baja, llevándolas a zonas más altas. lo que causa la infección del parénquima, con cicatrización secundaria y pérdida de función renal. El aumento crónico del residuo vesical y de las presiones de vaciamiento (>40 cm H2O) produce una lesión hidrostática progresiva del riñón de la mayoría de los pacientes, incluso en ausencia de infección o reflujo.
El reflujo vesicoureteral puede manifestarse con dolor abdominal o en los flancos, infecciones urinarias persistentes o recidivantes, disuria o dolor de costado al orinar, polaquiuria y urgencia o con signos de insuficiencia renal. También pueden encontrarse piuria, hematuria, proteinuria y bacteriuria.
Las cistouretrografías de llenado y miccional demuestran el reflujo y son la técnica más adecuada para diagnosticar la obstrucción a la salida de la vejiga, susceptible de reparación quirúrgica. Enla PIV pueden encontrarse una dilatación de los cálices, un «encintamiento» ureteral y ureterectasias con dilatación del sistema colector superior. La cistoscopia isotópica (con sonda) también puede revelar el reflujo. En los casos indicados, las escintigrafías con succimero (ácido dimercaptosuccínico) permiten estudiar la afectación de la corteza renal por infecciones agudas o cicatrices.
El reflujo vesicoureteral suele ser leve o moderado (con dilatación calicial escasa o nula) y suele ceder espontáneamente al cabo de algunos meses a varios años, siempre que se mantenga una profilaxis antibacteriana diaria. La mejor manera de tratar las infecciones, que se producen a pesar de la profilaxis, o la cicatrización renal progresiva consiste en la reimplantación de los uréteres. Cuando el reflujo va acompañado de orina residual o al vaciamiento vesical con alta presión, el tratamiento consiste en reducir la presión vesical con farmacoterapia, métodos conductuales o ambos. De esta forma ceden algunos reflujos pero, en caso contrario, se optará por la reimplantación. Con ella se elimina casi siempre el reflujo y se reducen al mínimo las pielonefritis futuras, con la consiguiente disminución de la morbilidad y la mortalidad por enfermedad renal secundaria al reflujo y la infección.
VEJIGA
Las anomalías congénitas de la vejiga son la extrofia, la agenesia, la duplicación, la persistencia del uraco, el síndrome de megavejiga, que podría ser un defecto mioneural primario (v. también Síndrome de megavejiga, cap. 216), y los divertículos.
Extrofia. Es una anomalía importante y grave, fácilmente detectable, en la que la vejiga aparece abierta (descubierta) en la región suprapúbica, observándose cómo la orina gotea a partir de los meatos ureterales. La mucosa de la vejiga se continúa con la piel del abdomen y los huesos pubianos están separados. El pronóstico en cuanto al mantenimiento de una función renal normal es bueno. En casi todos los casos es posible reconstruir la vejiga y devolverla a la pelvis, aunque siempre existirá reflujo vesicoureteral. Para tratar los reservorios vesicales que no se expanden suficientemente o que muestran una insuficiencia del esfínter, puede recurrirse a la ureterosigmoidostomía o a otros tipos de derivaciones urinarias continuas. Es necesario reconstruir los genitales.
Divertículos congénitos de la vejiga. Este tipo de divertículos predisponen a la infección urinaria y pueden asociarse a reflujo. El diagnóstico se hace por cistografía y cistoscopia. Puede estar indicada la extirpación quirúrgica de los divertículos con reconstrucción de la vejiga.
PENE Y URETRA
Existen casos de ausencia congénita del pene en el varón y de ausencia de la uretra tanto en el varón como en la mujer. Otras anomalías son el hipospadias, el epispadias, el pene doble, la incurvación o malrotación peneana congénita, el microfalo, las válvulas, estenosis, estrecheces o duplicaciones uretrales, la estenosis del meato y la fimosis y parafimosis.
Hipospadias. El desplazamiento del meato uretral se debe a un fallo en la tunelización y fusión del surco uretral. En el hipospadias de la mujer, la uretra se abre en el introito vaginal. En el varón, el prepucio no forma una circunferencia y aparece como una caperuza dorsal. La apertura de la uretra puede localizarse en el lado inferior del tallo peneano, en la unión penoescrotal, entre los pliegues del escroto o en el perineo. El «encordamiento» (incurvación ventral del pene, más evidente en la erección), suele asociarse a hipospadias y se debe a una banda fibrosa que sigue el trayecto normal del cuerpo esponjoso. El pronóstico en cuanto a la corrección funcional y estética es bueno. Durante el primer año de la vida puede repararse el encordamiento y reconstruir una neouretra utilizando piel del tallo del pene o del prepucio.
Epispadias. Es un defecto de fusión dorsal de la uretra que puede ser parcial (15% de los casos) o completo; la forma más grave se asocia a extrofia vesical. Es más frecuente en varones. En el epispadias parcial el control urinario puede ser satisfactorio. La reconstrucción aislada del pene puede lograr una continencia persistente, siendo a menudo necesario reconstruir también el tracto de salida de la vejiga para que el paciente desarrolle un control urinario eficaz.
Válvulas uretrales. Los pliegues congénitos que ocurren en la uretra prostática actúan como válvulas (válvulas uretrales posteriores). Las complicaciones se deben a la obstrucción, que puede ser intensa y provocar una alteración funcional miógena de la vejiga con alteraciones importantes de la vía superior e insuficiencia renal. Los síntomas y signos son un chorro urinario débil y goteante, incontinencia por rebosamiento e IU. El diagnóstico se establece mediante la cistouretrografía de vaciamiento. El tratamiento inicial es endoscópico. La cirugía precoz puede evitar el deterioro renal progresivo. Existe otra entidad, el divertículo de la uretra anterior (válvula uretral anterior), que también puede actuar como una válvula y cuyo tratamiento es asimismo endoscópico.
Estenosis uretral. Aunque la estenosis uretralen el varón es adquirida la mayor parte de lasveces, típicamente debida a una lesión por aplastamiento en un traumatismo de tipo «silla demontar» (v. también Traumatismos uretrales, cap.232), puede ser congénita. La estenosis adquirida puede manifestarse por un exudado sanguinolento posvaciamiento y puede resolverse espontáneamente o progresar hacia una estrechez verdadera. El tratamiento inicial consiste en una uretrotomía endoscópica.
Estenosis del meato uretral. La estenosis del meato uretral asociada a hipospadias es la forma más frecuente de estenosis congénita de la uretra, aunque en la mayor parte de los casos se trata de un cuadro adquirido secundario a la cicatrización de una irritación del meato en niños que son circuncidados mientras aún llevan pañales. Si el chorro de orina muestran una desviación significativa o es puntiforme, estará indicada la meatotomía.
Fimosis y parafimosis. La fimosis consiste en una estrechez congénita o adquirida (inflamatoria) del prepucio que impide su retracción. La parafimosis es la imposibilidad de reducir por encima del glande un prepucio retraído que produce constricción. En ambas circunstancias está indicada la circuncisión, pudiendo ser necesario practicar una incisión dorsal preliminar. El pronóstico es excelente.
TESTÍCULOS Y ESCROTO
Las alteraciones de desarrollo del escroto pueden ser unilaterales o bilaterales y a menudo se asocian a criptorquidia. Los hemangiomas congénitos del escroto pueden sangrar o aumentar progresivamente de tamaño, obligando a su extirpación quirúrgica. El hidrocele congénito comunica con la cavidad abdominal por una prolongación vaginal persistente, un espacio herniario potencial, pero pueden curar espontáneamente si la comunicación se oblitera durante la lactancia. Los hidroceles comunicantes que persisten después de los 9a 12 meses suelen precisa la reparación quirúrgica. Una anomalía estéticamente llamativa pero corregible con cirugía es la trasposición penescrotal, en la que el escroto ocupa una posición más cefálica de la habitual, y el pene una más caudal.
Criptorquidia (testículos no descendidos). En alrededor del 1% de los varones, el descenso de uno o ambos testículos es incompleto o insuficiente, La función hormonal suele ser normal pero, si no se trata, la criptorquidia puede dar lugar a una insuficiencia progresiva de la espermatogénesis y se asocia a una elevada incidencia de carcinoma, que aparece en la adolescencia o en la vida adulta.
El testículo verdaderamente no descendido permanece en la cavidad abdominal o en el retroperitoneo debido a mecanismos hormonales o mecánicos. Cuando el descenso es incompleto (parada o maldescenso), el testículo se encuentra en el canal inguinal y la causa suele ser una obstrucción mecánica. Los testículos ectópicos se encuentran fuera del trayecto habitual de descenso, en situación suprapúbica, en el perineo o a lo largo de la cara interna del muslo. Los testículos hipermóviles o retráctiles pueden alcanzar el escroto (p. ej., durante un baño caliente), pero vuelven a retraerse hacia el canal inguinal. Algunos testículos ectópicos o con descenso incompleto sólo se manifiestan cuando prosigue el crecimiento lineal del cuerpo, revelándose su sutil malposición porque el testículo permanece fijo cerca del anillo inguinal externo, en lugar de reposar en el escroto.
Los testículos hipermóviles (retráctiles) no precisan tratamiento quirúrgico, siempre que el cordón espermático sea lo bastante largo como para permitir que el testículo descanse en una posición escrotal declive sin tracción (cuando el reflejo cremastérico está inactivo). La hipermotilidad suele resolverse de forma espontánea durante la adolescencia. La administración de gonadotropina coriónica humana en dosis de 250 a 1.000 UI i.m. 2 o 3 veces a la semana durante un máximo de 6 sem puede facilitar el descenso testicular bilateral. Las dosis recomendadas son 250 UI para los lactantes de 3 a 12 meses, 500 UI para los niños de 1 a 6 años y 1.000 UI para los >6 años, con dosis máximas totales de 2.500, 5.000 y 10.000 UI, respectivamente. El tratamiento quirúrgico del testículo no descendido suele consistir en la orquiopexia, que se efectúa hacia el año de vida. El retraso de la cirugía hasta después de los 2años implica una alteración de la espermatogénesis, lo que resulta crítico si la criptorquidia es bilateral. La orquidectomía es el tratamiento de elección cuando se descubre una criptorquidia unilateral en un paciente pospuberal.
Torsión testicular. La rotación de un testículo sobre su cordón, de forma espontánea o tras un traumatismo, puede dar lugar a un desarrollo anómalo de la túnica vaginal y del cordón espermático. Los síntomas inmediatos son un intenso dolor local, náuseas y vómitos, seguidos de edema e induración escrotal. La torsión debe diferenciarse de los cuadros inflamatorios que afectan al contenido del escroto y de los tumores testiculares; la escintigrafía isotópica escrotal o la ecografía Doppler en color pueden establecer el diagnóstico. Cuando la exploración inicial no permite descartar la torsión, se recomienda una intervención inmediata sin esperar a los estudios radiológicos, ya que sólo una exploración realizada en las primeras horas ofrecerá probabilidades de salvar el testículo. En la intervención se procede a la fijación del testículo contralateral para evitar su torsión (v. también en Exploración física completa, cap. 256).
ANOMALÍAS DEL TRANSPORTE TUBULAR RENAL
CISTINURIA
Defecto hereditario de los túbulos renales consistente en la alteración de la reabsorción del aminoácido cistina, con aumento de su excreción urinaria y formación de cálculos de cistina en la vía urinaria.
La cistinuria se transmite de forma autosómica recesiva. Los heterocigotos pueden excretar cantidades superiores a las normales de cistina en la orina, pero rara vez en magnitud suficiente para la formación de cálculos.
Fisiopatología
El defecto primario es una disminución de la reabsorción tubular renal de cistina con aumento de su concentración en la orina. La cistina es poco soluble en la orina ácida y, cuando su concentración supera su umbral de solubilidad, precipita, con formación tanto de cálculos como de cristales.
También existe una alteración de los aminoácidos dibásicos (lisina, ornitina y arginina), aunque esto no produce problemas, ya que éstos disponen de un sistema de transporte alternativo distinto del que comparten con la cistina. Además, su solubilidad urinaria es mayor que la de esta última y el aumento de su excreción no da lugar a la formación de cristales ni de cálculos. Por otra parte, disminuye su absorción en el intestino delgado (también la de la cistina).
Síntomas, signos y diagnóstico
Los síntomas, de los que el más frecuente es elcólico renal, suelen aparecer entre los 10 y 30años. La obstrucción puede facilitar las UI y la insuficiencia renal.
Se forman cálculos radiopacos de cistina en la pelvis renal o en la vejiga, a menudo con morfología en asta de ciervo. La cistina puede aparecer en la orina en forma de cristales hexagonales de color pardo-amarillento. El exceso urinario de cistina puede detectarse mediante la prueba del cianuro y nitroprusiato. El diagnóstico se confirma por cromatografía o electroforesis.
Pronóstico y tratamiento
Los pacientes que sobreviven durante muchos años suelen desarrollar una nefropatía terminal. La concentración urinaria de cistina puede reducirse aumentando el volumen de orina. La ingesta de líquidos debe ser suficiente para que la diuresis llegue a unos 4 l/d. La hidratación es importante por la noche, cuando el pH de la orina desciende. La alcalinización de la orina hasta un pH >7,5 mediante la administración de bicarbonato sódico oral en varias tomas y acetazolamida en dosis de 5mg/kg (hasta 250 mg) oral en el momento de acostarse aumentan significativamente la solubilidad de la cistina. Si la elevada ingesta de líquidos y la alcalinización no reducen la formación de cálculos, pueden intentarse otras medidas, como la administración de penicilamina, aunque su toxicidad limita su utilidad. La mitad de todos los pacientes tratados con penicilamina desarrollan alguna manifestación tóxica, como fiebre, erupción, artralgias y, con menos frecuencia, síndrome nefrótico, pancitopenia o una reacción de tipo LED.
SÍNDROME DE FANCONI
Trastorno hereditario o adquirido, a menudo asociado a cistinosis, con anomalías características de la función tubular proximal renal como glucosuria, fosfaturia, aminoaciduria y pérdida de bicarbonato.
Etiología y epidemiología
El síndrome de Fanconi hereditario suele acompañar a otros trastornos genéticos, especialmente a la cistinosis, y se transmite de forma autosómica recesiva (v. tabla 269-5). Los heterocigotos pueden presentar acumulación de cistina en las células pero sin otras manifestaciones clínicas ni analíticas. También puede acompañar a la enfermedad de Wilson, a la intolerancia hereditaria a la fructosa, a la galactosemia, a la glucogenosis, al síndrome de Lowe o a la tirosinemia.
El síndrome de Fanconi adquirido puede deberse a la administración de 6-mercaptopurina o de tetraciclina caducada, al trasplante renal, al mieloma múltiple, a la amiloidosis, a la intoxicación por metales pesados u otros agentes químicos o a la deficiencia de vitamina D.
Fisiopatología, síntomas y signos
Existen diversos defectos de la función tubular proximal renal como reabsorción alterada de la glucosa, los fosfatos, los aminoácidos, el bicarbonato, el ácido úrico, el agua, el potasio y el sodio. Hay una aminoaciduria generalizada y, a diferencia de lo que sucede en la cistinuria, el aumento de la excreción de cistina es sólo un componente menor. Se desconoce cuál es la anomalía fisiopatológica básica.
En el síndrome de Fanconi hereditario, las manifestaciones clínicas más importantes (acidosis tubular proximal, raquitismo hipofosfatémico, hipocaliemia, poliuria, polidipsia) aparecen habitualmente durante la lactancia.
En la forma nefropática asociada a cistinosis, es frecuente el retraso del crecimiento y desarrollo. Las retinas muestran una despigmentación parcheada. Hay también una nefritis intersticial causante de una insuficiencia renal progresiva que puede ser fatal antes de que el paciente alcance la adolescencia.
En el síndrome de Fanconi adquirido, los adultos muestran alteraciones analíticas propias de la acidosis tubular renal (de tipo proximal), hipofosfatemia e hipocaliemia. Pueden observarse síntomas de enfermedad ósea (osteomalacia) y debilidad muscular.
Diagnóstico y tratamiento
El diagnóstico se hace mediante la demostración de las alteraciones de la función renal, especialmente la glucosuria, la fosfaturia y la aminoaciduria. En la cistinosis, la exploración con lámpara de hendidura revela la presencia de cristales de cistina en la córnea.
Salvo por la eliminación de la nefrotoxina responsable en los casos en que exista, no hay tratamiento específico alguno. La administración oral de bicarbonato sódico puede reducir la acidosis. La solución de Shol en dosis de 2 a 3 mEq/kg/d o 5 a 15 ml tras las comidas y al acostarse, Bicitra oPolycitra-K, pueden sustituir a la solución de bicarbonato cálcico, siendo mejor toleradas. Puede ser necesario aportar K suplementario. El raquitismo hipofosfatémico se tratará de la forma descrita más adelante. En la insuficiencia renal, el trasplante resulta satisfactorio, pero cuando la enfermedad subyacente es una cistinosis, la lesión progresiva puede continuar en otros órganos y acabar con la vida del paciente.
RAQUITISMO HIPOFOSFATÉMICO
(Raquitismo resistente a la vitamina D)
Trastorno familiar o, raras veces, adquirido que se caracteriza por hipofosfatemia, absorción intestinal defectuosa del calcio y raquitismo u osteomalacia que no responden a la administración de vitamina D.
El raquitismo hipofosfatémico familiar se hereda de forma dominante ligado al cromosoma X. La afectación ósea es menor en las mujeres que en los varones, pudiendo causar tan sólo hipofosfatemia. A veces, los casos esporádicos adquiridos se asocian a tumores mesenquimales benignos (raquitismo oncogénico).
Fisiopatología
La principal anomalía fisiopatológica es la disminución de la reabsorción tubular renal proximal de fosfato, a la que se une una menor absorción intestinal de calcio y fósforo. Los niveles de vitamina D y de hormona paratiroidea son normales.
Existen dos tipos de raquitismo hipofosfatémico. En el tipo I, la alteración de la síntesis renal hace que el nivel plasmático de 1,25-dihidroxivitamina D3 sea bajo. En el tipo II, la 1,25-dihidroxivitamina D3 es normal o está elevada y la enfermedad se debe a una alteración de la respuesta celular a dicha sustancia.
Síntomas, signos y diagnóstico
La enfermedad se manifiesta por un espectro de alteraciones que van desde la hipofosfatemia aislada hasta un raquitismo u osteomalacia graves con incurvación de las piernas y otras deformidades óseas, seudofracturas, dolor óseo y talla corta. Las excrecencias óseas en las inserciones de los músculos limitan el movimiento. El raquitismo de la columna o de la pelvis que se produce en la deficiencia dietética de vitamina D es raro en la forma hipofosfatémica en la que, por otra parte, sí pueden encontrarse craneostenosis y convulsiones; la edad de comienzo suele ser anterior al primer año de vida.
En la sangre se encuentran hipofosfatemia con calcio normal y fosfatasa alcalina a menudo elevada. Los niveles séricos de 1,25-dihidroxivitamina D3 permiten diferenciar los tipos I y II. Cuando se interpretan estos niveles, debe tenerse en cuenta el fosfato sérico que existía en el momento en que se tomó la muestra de sangre. Un fosfato bajo debe elevar el nivel de 1,25-dihidroxivitamina D3. El raquitismo hipofosfatémico debe diferenciarse del raquitismo dependiente de la vitamina D, una enfermedad autosómica recesiva de características clínicas similares aunque en ella existe hipocalcemia, la hipofosfatemia es escasa o nula, la tetania y las convulsiones son frecuentes y provoca raquitismo de la columna y de la pelvis (v. también cap. 3).
Tratamiento
El tratamiento del raquitismo hipofosfatémico tipo I consiste en administración de fosfato oral en dosis de 1 a 3 g/d en tomas fraccionadas, en forma de solución de fosfato neutro, más calcitriol (1,25-dihidroxivitamina D3) 0,015 a 0,02 mg/ kg/d como dosis inicial y 0,03 a 0,06 mg/kg/d hasta un máximo de 2 mg/d, como mantenimiento. Con ello se logra incrementar las concentraciones plasmáticas de fosfato y disminuir las de fosfatasa alcalina, curar el raquitismo y mejorar la velocidad de crecimiento. El tratamiento puede verse complicado por hipercalcemia, hipercalciuria y reducción de la función renal. El raquitismo hipofosfatémico de tipo II responde mal al tratamiento y sólo se hace un aporte de fósforo para reducir en lo posible el grado de hipofosfatemia. En los adultos con raquitismo oncogénico se han obtenido mejorías espectaculares al extirpar el tumor mesenquimatoso de células pequeñas que produce un factor humoral inhibidor de la reabsorción de fosfatos en el túbulo renal proximal.
ENFERMEDAD DE HARTNUP
Enfermedad rara debida a absorción y excreción anormales de tritófano y de otros aminoácidos y que clínicamente se caracteriza por erupción cutánea y alteraciones del SNC.
La enfermedad de Hartnup se hereda de forma autosómica recesiva, siendo la consanguinidad un factor etiológico frecuente. Los heterocigotos son fenotípicamente normales.
En esta enfermedad existe malabsorción de tritófano, fenilalanina, metionina y otros aminoácidos monoaminomonocarboxílicos en el intestino delgado. La acumulación de los aminoácidos no absorbidos en el aparato digestivo provoca un aumento de su metabolismo por la flora intestinal, lo que hace que se absorban algunos productos de la degradación del triptófano como derivados indol, quinurenina y serotonina, que aparecen en la orina. Existe también un defecto en la reabsorción renal de los aminoácidos, lo que se traduce en una aminoaciduria generalizada que afecta a todos los aminoácidos neutros, salvo la prolina y la hidroxiprolina. Por último, la conversión de triptófano en niacinamida es también defectuosa.
La aparición de los síntomas va precedida casi siempre de alteraciones de la ingesta nutritiva. Los síntomas y signos se deben a la deficiencia de niacinamida y son similares a los de la pelagra, sobre todo por la erupción que surge en las partes del cuerpo expuestas a la luz solar. Las manifestaciones neurológicas consisten en ataxia cerebelosa y anomalías psicológicas, siendo frecuentes el retraso mental, la talla corta, las cefaleas y los colapsos o desvanecimientos. Aunque la enfermedad existe ya al nacimiento, los síntomas suelen manifestarse durante la lactancia, la infancia o incluso al comienzo de la edad adulta. La luz del sol, la fiebre, algunos fármacos y otros tipos de estrés pueden desencadenar la sintomatología.
El diagnóstico se hace por el característico patrón de excreción de aminoácidos en la orina. El hallazgo en ésta de indoles y de otros productos de la degradación del triptófano constituye una prueba adicional de la enfermedad. El pronóstico es bueno y la frecuencia de los ataques suele disminuir con la edad. Es posible prevenir los ataques manteniendo una buena nutrición y aportando suplementos de niacinamida o niacina a la dieta (20 mg/kg, v.o.). Los ataques pueden tratarse con nicotinamida (50 a 400 mg/d, v.o.).
IMINOGLICINURIA FAMILIAR
Defecto autosómico recesivo benigno de la reabsorción tubular renal de iminoácidos y de glicina.
Los homocigotos excretan cantidades anormales de iminoácidos (prolina e hidroxiprolina) y de glicina; los heterocigotos sólo presentan glicinuria. Los niveles plasmáticos de los aminoácidos son normales. La absorción intestinal de prolina puede estar alterada. La iminoaciduria es normal en el RN.
ANOMALÍAS CROMOSÓMICAS
Los cromosomas son las 46 estructuras alargadas que se observan durante la división celular en el núcleo de la mayoría de las células humanas. Están formados por proteínas y ADN y en ellos se encuentran la mayoría de los genes (unidades de herencia; v. también cap. 286).
Diagnóstico
La preparación de los cromosomas para su análisis es relativamente sencilla y para ello suelen utilizarse linfocitos circulantes; en el caso del feto, se recurre a amniocitos del líquido amniótico o a células de las vellosidades coriónicas de la placenta. Las células se cultivan en el laboratorio con fitohemaglutinina para estimular su división. A continuación, se añade colchicina para detener las mitosis durante la metafase, momento en que los cromosomas se han replicado en dos cromátides unidas a los centrómeros. Las células, extendidas en portaobjetos, se tiñen, se fotografían los cromosomas de una célula, se recortan las imágenes obtenidas y se pegan en un papel para obtener el cariotipo. Para producir una muestra visual de los cromosomas puede utilizarse también una imagen de ordenador.
La tinción de los cromosomas se hace con técnicas de bandeo G (Giemsa) o Q (fluorescencia). Otras técnicas adicionales de tinción y para la extensión de los cromosomas en toda su longitud han mejorado mucho la precisión del diagnóstico citogenético (v. fig. 261-9).
Las nuevas técnicas moleculares utilizan sondas de ADN (que pueden tener apéndices fluorescentes) para localizar genes o secuencias de ADN concretas en los cromosomas. La hibridación in situ con fluorescencia (FISH, fluorescent in situ hybridization) se emplea para identificar la organización de los genes y buscar deleciones, reordenamientos y duplicaciones cromosómicas.
La nomenclatura del cariotipo es la siguiente: el varón normal se designa como 46,XY y la mujer normal, como 46,XX. En el síndrome de Down debido a la existencia de un cromosoma 21 accesorio (trisomía 21) se utilizan las anotaciones47,XY,21+ para el varón y 47,XX,21+ para lamujer. En el síndrome de Down debido a unatraslocación (dos cromosomas que han unido), la típica madre «portadora de traslocación equilibrada» 14/21 se representa como 45,XX, t(14q;21q). El cromosoma translocado (t) se forma a partir de 14q y 21q ([q] representa el brazo largo; los brazos cortos [p] se han perdido). En el caso de una deleción del brazo corto de un cromosoma 5 (como sucede en el síndrome de deleción 5p), el cariotipo femenino será 46,XX,5p-.
Según su longitud, cada brazo de un cromosoma se divide en una a cuatro regiones principales; cada banda, con tinción positiva o negativa, recibe un número, que es mayor a medida que crece su distancia respecto al centrómero. Por ejemplo, 1q23 designa al cromosoma (1), al brazo largo (q), a la segunda región distal al centrómero (2) y a la tercera banda (3) de esa región (v. fig. 261-9).
ANOMALÍAS AUTOSÓMICAS
Las trisomías completas, las parciales (debidas a translocaciones de porciones de brazos cortos o largos) y las deleciones de diversos cromosomas dan lugar a muchos síndromes distintos, de los que aquí sólo se tratarán los más frecuentes en la clínica.
Síndrome de Down
(Trisomía 21; trisomía G; mongolismo)
Anomalía cromosómica que suele manifestarse con retraso mental, una facies característica y muchas otras manifestaciones típicas, entre ellas microcefalia y talla corta.
La incidencia global de la trisomía 21 en los RN vivos es de alrededor de 1/800, pero existen grandes variaciones que dependen de la edad de la madre: en las mujeres <20 años, la incidencia es de 1/2.000, mientras que en las >40 años se eleva hasta alrededor de 1/40 (v. también tabla 247-1). Aproximadamente el 20% de los niños con síndrome de Down son hijos de madres >35 años. El síndrome puede ser consecuencia de una trisomía21, de una traslocación o de un mosaicismo.
Trisomía 21. En el 95% de los casos existe un cromosoma 21 completo accesorio, que en el 95% de los afectados procede de la madre.
Traslocación (t). Algunos pacientes con síndrome de Down tienen 46 cromosomas pero realmente poseen el material genético de 47; el cromosoma 21 adicional se encuentra translocado o unido a otro cromosoma.
La traslocación más frecuente es t(14;21), en la que el cromosoma 21 adicional está unido a un cromosoma 14. En la mitad de los casos, los cariotipos de los dos padres son normales, lo que indica una traslocación de novo en el niño afectado. En la otra mitad, uno de los padres (casi siempre la madre), aunque fenotípicamente normal, sólo tiene 45 cromosomas, uno de los cuales es t(14;21). Teóricamente, la probabilidad de que una madre portadora tenga un hijo con síndrome de Down es de 1:3 pero por, razones desconocidas, el riesgo real es inferior (alrededor de 1:10); si el portador es el padre, el riesgo será sólo 1:20.
La siguiente traslocación en frecuencia es t(21;22). El riesgo de que una madre portadora tenga un hijo con síndrome de Down es de 1:10, riesgo que será aún menor si el portador es el padre. En casos extraordinariamente raros, uno de los progenitores es portador de t(21;21). En estos casos, el 100% de los hijos supervivientes tendrá un síndrome de Down.
Mosaicismo. El mosaicismo ocurre cuando una persona tiene dos líneas celulares distintas. En el síndrome de Down es probable que el mosaicismo se deba a un error en la separación de los cromosomas en la división celular (no disyunción) en el embrión en desarrollo. La mayoría de los pacientes poseen dos líneas celulares, una normal y la otra con 47 cromosomas. La proporción relativa de cada línea celular es muy variable, tanto de unas personas a otras como dentro de los distintos tejidos y órganos de una misma persona. El pronóstico en cuanto a la inteligencia depende de la proporción de células con trisomía 21 presentes en el cerebro. Se han identificado algunas personas con síndrome de Down de tipo mosaico con signos clínicos apenas reconocibles e inteligencia normal. La incidencia del mosaicismo en este síndrome es desconocida. Si uno de los progenitores tiene un mosaicismo para trisomía 21 en la línea germinal, el riesgo de tener un segundo hijo afectado es mayor.
Síntomas y signos
Los RN tienden a ser plácidos, rara vez lloran y muestran hipotonía muscular. Es frecuente que presenten un exceso de piel alrededor del cuello, lo que puede detectarse en la ecografía fetal como edema de esa región. Existe un retraso del desarrollo tanto físico como mental; el cociente de inteligencia medio (CI) es de alrededor de 50.
Es característico que los pacientes muestren microcefalia, un occipucio plano y una talla corta. La parte externa de los ojos muestra una hendidura dirigida hacia arriba y en el ángulo interno del ojo suelen encontrarse pliegues epicánticos. Las manchas de Brushfield (manchas grises o blanquecinas que parecen granos de sal alrededor de la periferia del iris), que suelen encontrarse al nacimiento, desaparecen durante los primeros 12 meses de vida. El puente nasal es plano, la boca se mantiene abierta debido a una lengua larga que protruye, con un aspecto plegado y sin surco central, mientras que la orejas son pequeñas y redondeadas. Las manos son cortas y anchas y a menudo sólo tienen un pliegue palmar (pliegue simiesco); los dedos son cortos, con clinodactilia (incurvación) del 5.º dedo, que a menudo tiene sólo dos falanges. El pie puede tener una amplia hendidura entre los dedos 1.º y 2.º y un surco plantar suele extenderse hacia atrás sobre el pie. Las huellas dactilares de las manos y los pies son características (dermatoglifos).
Alrededor del 40% de los RN afectados padecen una cardiopatía congénita, de las que las más frecuentes son las que afectan a los tabiques inter-auricular e interventricular. Existe mayor incidencia de casi todas las demás malformaciones congénitas, especialmente de atresia duodenal.
Muchos pacientes con síndrome de Down desarrollan problemas de tiroides que pueden resultar difíciles de detectar, a menos que se hagan análisis de sangre. Además, muestran tendencia a desarrollar trastornos de la audición y la visión. Por ello, es adecuado establecer controles regulares.
En la autopsia, todos los cerebros de adultos con síndrome de Down muestran las características microscópicas típicas de la enfermedad de Alzheimer y muchas personas desarrollan los signos clínicos asociados a ella. Algunas mujeres afectadas son fértiles, siendo la probabilidad de que el feto tenga el síndrome de Down del 50%. Sin embargo, muchos de estos fetos afectados acaban en abortos espontáneos. Todos los varones con síndrome de Down son estériles.
Pronóstico
La esperanza de vida es inferior a la normal, debido a las cardiopatías y a la tendencia a desarrollar una leucemia aguda. Casi todos los pacientes sobreviven hasta la edad adulta, en la que parece acelerarse el proceso de envejecimiento, de manera que la muerte suele sobrevenir en el quinto o sexto decenios.
Trisomía 18
(Síndrome de Edwards; trisomía E)
Alteración cromosómica debida a un cromosoma18 accesorio y que se caracteriza por múltiples anomalías del desarrollo, entre ellas un retraso mental grave.
La trisomía 18 afecta a 1 de cada 6.000 RN vivos, aunque muchas de estas concepciones terminan en aborto espontáneo. Más del 95% de los niños afectados tienen una trisomía 18 completa y el material cromosómico adicional procede de la madre en el 95% de los casos. El riesgo aumenta a medida que lo hace la edad de la madre. Por cada varón nacido con trisomía 18, nacen tres mujeres.
El RN es muy pequeño para su edad gestacional y muestra hipotonía e hipoplasia importante del músculo esquelético y de la grasa subcutánea. El llanto es débil, con disminución de la respuesta a los sonidos. A menudo existe una historia de actividad fetal escasa, polihidramnios, una placenta pequeña y una sola arteria umbilical. Los rebordes orbitarios son hipoplásicos, las fisuras palpebrales son cortas y la boca y la mandíbula, pequeñas, por lo que la cara del niño un aspecto demacrado. Son frecuentes la microcefalia, el occipucio prominente, las orejas de implantación baja y un esternón corto. Suele observarse un puño cerrado peculiar con el dedo índice colocado sobre los dedos tercero y cuarto. Falta el pliegue distal del quinto dedo y el patrón del reborde dérmico de las puntas de los dedos muestra un arco bajo. Las uñas de las manos son hipoplásicas y el dedo gordo del pie es corto y a menudo se encuentra en flexión dorsal. Son frecuentes los pies zambos y en «mecedora». En el síndrome se encuentran diversas cardiopatías congénitas y malformaciones de los pulmones, diafragma, pared abdominal, riñones y uréteres. Además, son asimismo frecuentes las hernias, la diastasis de los rectos, la criptorquidia y pliegues cutáneos redundantes (sobre todo, en la cara posterior del cuello).
Es raro que el niño sobreviva más de algunos meses y menos del 10% superan el año de vida. Los supervivientes sufren un importante retraso del desarrollo y numerosas discapacidades.
Trisomía 13
(Síndrome de Patau, trisomía D)
Alteración cromosómica debida a la existencia de un cromosoma 13 adicional y caracterizado por muchas alteraciones del desarrollo, entre ellas retraso mental grave y malformaciones del encéfalo anterior.
La trisomía 13 afecta aproximadamente a 1 de cada 10.000 nacidos vivos, y de ellos el 80% presentan una trisomía completa. El riesgo aumenta a medida que lo hace la edad materna y el material cromosómico adicional suele proceder de la madre.
En este síndrome son características las anomalías de la línea media y son frecuentes los defectos anatómicos macroscópicos del encéfalo, sobre todo la holoprosencefalia (división incompleta del encéfalo anterior), el labio leporino, el paladar hendido, la microftalmia, los colobomas (fisuras) del iris y la displasia retiniana. Los rebordes orbitarios son planos y las fisuras palpebrales suelen tener forma de hendidura. La morfología de las orejas es anómala y su implantación es baja. Es frecuente la sordera. Los RN tienden a ser pequeños para su edad gestacional. Se observan pliegue simiesco, polidactilia y uñas estrechas y excesivamente convexas. Alrededor del 80% de los pacientes presentan graves cardiopatías congénitas, siendo frecuente la dextrocardia. Otras anomalías posibles de la línea media son los defectos del cuero cabelludo y los senos dérmicos. En la parte posterior del cuello suelen existir pliegues cutáneos laxos. Los genitales suelen ser anormales en ambos sexos: en los niños suele haber criptorquidia y un escroto anormal y las niñas tienen un útero bicorne. En las primeras etapas de la vida son frecuentes las crisis de apnea. El retraso mental es grave.
La mayoría de los pacientes (70%) muestran una afectación tan grande que mueren antes de los 6 meses, de forma que el porcentaje de los que sobreviven más de 1 año no alcanza el 10%.
Síndromes de deleción
Síndromes clínicos debidos a la pérdida de partes de cromosomas.
Deleción 5p (síndrome del maullido de gato). Consiste en la deleción del extremo del brazo corto del cromosoma 5 y se caracteriza por un llanto agudo, muy parecido al maullido de un gato pequeño; este llanto se mantiene durante algunas semanas pero luego desaparece. Los pacientes son de bajo peso para su edad gestacional y presentan microcefalia, simetría facial, cara redonda con ojos separados, fisuras palpebrales antimongoloides o dirigidas hacia abajo con o sin pliegues epicánticos, estrabismo y nariz de base ancha. Las orejas son de implantación baja y forma anormal, con conductos auditivos externos estrechos y colgajos preauriculares. Se observa sindactilia de grado diverso; las cardiopatías congénitas son frecuentes y los niños se hallan hipotónicos. Existe un importante retraso físico y mental. Muchos sobreviven hasta la edad adulta, pero con grandes discapacidades.
Deleción 4p (síndrome de Wolf-Hirschhorn). Este síndrome se caracteriza por un retraso mental profundo. Además, existen una nariz ancha o en pico, defectos en la línea media del cuero cabelludo, ptosis y colobomas, paladar hendido, retraso de la edad ósea y, en los varones, hipospadias y criptorquidia. La tasa de mortalidad es elevada durante la lactancia; los supervivientes, relativamente escasos, tienden a desarrollar infecciones y epilepsia. Los pocos que sobreviven hasta el tercer decenio muestran graves minusvalías.
Los síndromes de genes contiguos consisten en microdeleciones y deleciones submicroscópicas de genes contiguos en partes concretas de muchos cromosomas. Pueden detectarse con sondas fluorescentes y con técnicas de extensión de los cromosomas. En la tabla 261-4 se recogen los más frecuentes. A menudo resulta imposible demostrar las deleciones con técnicas de citogenética, pero puede confirmase su existencia con sondas de ADN específicas del área perdida.

Las deleciones teloméricas (deleciones de cualquiera de los extremos de un cromosoma) son la causa de muchos de los casos inespecíficos de retraso mental con características dismórficas.
ANOMALÍAS DE LOS CROMOSOMAS SEXUALES
En el ser humano, la determinación del sexo está controlada por los cromosomas X e Y. La mujer tiene dos cromosomas X y los varones un cromosoma X y otro Y. El cromosoma Y es uno de los más pequeños del cariotipo y parece que su función principal está relacionada con la determinación del sexo masculino. Por el contrario, el cromosoma X es uno de los cromosomas más grandes y contiene miles de genes que, en su mayoría, no tienen relación alguna con la determinación del sexo.
Hipótesis Lyon (inactivación de X). La mujer normal tiene dos loci por cada uno de los genes del cromosoma X, mientras que el varón sólo tiene uno. Aparentemente, ello podría dar lugar a un problema de «dosificación» genética. Sin embargo, según la hipótesis Lyon, uno de los dos cromosomas X de cada célula somática de la mujer se inactiva genéticamente al comienzo de la vida embrionaria. El cuerpo de Barr, o masa de cromatina sexual existente en el interior del núcleo de las células somáticas femeninas, corresponde al segundo cromosoma X inactivado. Se ha identificado el gen responsable de la inactivación de los genes del cromosoma X (XIST). Los estudios degenética molecular demuestran que no todos los genes del segundo cromosoma X se inactivan, aunque sí la gran mayoría. De hecho, cualquiera que sea el número de cromosomas X presentes en el genoma, todos, salvo uno, tienen casi todos sus genes inactivados.
La inactivación de X tiene interesantes implicaciones clínicas. Por ejemplo, las anomalías del cromosoma X son relativamente benignas en comparación con otras anomalías autosómicas análogas. Las mujeres con tres cromosomas X suelen ser física y mentalmente normales y son fértiles. Por el contrario, los efectos de todas las trisomías autosómicas conocidas son devastadores. De la misma forma, la ausencia de un cromosoma X, aunque se traduce en un síndrome específico (síndrome de Turner), es relativamente benigna; la ausencia de un autosoma es invariablemente letal.
La inactivación de X explica también el estado de portadora asintomática de los trastornos recesivos ligados a X. Las mujeres heterocigóticas para la hemofilia o para la distrofia muscular suelen ser asintomáticas, aunque en ocasiones muestren cierta tendencia a la hemorragia o a la debilidad muscular, respectivamente. La hipótesis Lyon sugiere que la inactivación de X es aleatoria, de forma que en cada persona se inactivarían el 50% del cromosoma X paterno y el 50% del materno. Sin embargo, los procesos aleatorios siguen la curva de distribución normal, por lo que en unas mujeres, en un tejido determinado, se encuentran inactivados los cromosomas X de origen paterno, y en otras los inactivos son los de origen materno. Si, por azar, en un tejido determinado de una heterocigota casi todas las células sufren la inactivación del alelo normal, esta persona manifestará una enfermedad similar a la de los varones afectados.
Síndrome de Turner
(Síndrome de Bonnevie-Ullrich)
Alteración de los cromosomas sexuales consistente en la ausencia parcial o completa de uno de los dos cromosomas sexuales, mostrando la paciente un fenotipo femenino.
El síndrome de Turner ocurre en alrededor de 1/4.000 RN vivos del sexo femenino. El 99% delas concepciones con 45,X acaba en aborto espontáneo. En el 80% de los RN vivos con monosomía X, el cromosoma perdido es el de origen paterno.
Las alteraciones cromosómicas son variables en las mujeres afectadas. Alrededor del 50% tienen un cariotipo 45,X. Muchas son mosaicos (p.ej., 45,X/46,XX o 45,X/47,XXX). El fenotipo varía entre el típico del síndrome de Turner y el normal. En ocasiones, las pacientes tienen un cromosoma X normal y otro en forma de anillo; para que esto suceda, es necesario que se pierda una pieza tanto del brazo corto como del brazo largo de un cromosoma X anormal. Algunas personas afectadas tienen un cromosoma X normal y un isocromosoma de brazo largo formado por la pérdida de los brazos cortos y el desarrollo de uncromosoma formado por dos brazos largos del cromosoma X. Estas personas tienden a mostrar muchas de las características fenotípicas del síndrome de Turner. Así pues, parece que la deleción del brazo corto del cromosoma X desempeña un papel importante en el fenotipo resultante.
Las recién nacidas afectadas suelen presentar un intenso linfedema en los dorsos de las manos y los pies y linfedema o pliegues cutáneos laxos en la nuca. Sin embargo, en muchas mujeres con síndrome de Turner la afectación es muy leve. Las características típicas consisten en talla corta, pliegues cutáneos en la nuca, ptosis, tórax ancho con pezones muy separados, múltiples nevos pigmentarios, cuartos metacarpianos y metatarsianos cortos, pulpejos prominentes con remolinos en los dermatoglifos de las puntas de los dedos delas manos, hipoplasia de las uñas, coartación de aorta, válvula aórtica bicúspide e hiperextensión del codo. Las malformaciones renales y los hemangiomas son frecuentes. A veces existen telangiectasias en el aparato GI que pueden dar lugar a hemorragias intestinales. La deficiencia mental es rara, pero muchas pacientes sufren cierta disminución de algunas capacidades perceptivas, lo que hace que su puntuación en los tests de inteligencia y en matemáticas sean bajas, incluso aunque las pruebas de inteligencia verbal sean normales o incluso superiores a la media. En el 90% de las personas afectadas se encuentra disgenesia gonadal con ausencia de pubertad, falta de desarrollo del tejido mamario y amenorrea. El tratamiento sustitutivo con hormonas femeninas produce la pubertad. Los ovarios están representados por unas cintillas bilaterales de estroma fibroso en las que no suelen existir óvulos. Sin embargo, entre el 5 y el 10% de las niñas afectadas desarrollan la menarquia espontáneamente y en ocasiones muy raras se han descrito casos de mujeres fértiles que han tenido hijos.
En todas las personas con disgenesia gonadal resulta necesario proceder a un estudio citogenético con sondas específicas de Y para descartarun mosaicismo en el que una de las líneas seaportadora de dicho cromosoma, por ejemplo, 45,X/46,XY. Estas personas suelen ser fenotípicamente mujeres con características variables de síndrome de Turner, en las que existe mayor riesgo de desarrollo de neoplasias malignas gonadales, sobre todo gonadoblastoma. En estos casos, debe procederse a la extirpación profiláctica de las gónadas tan pronto como se efectúe el diagnóstico.
Síndrome de X triple (47,XXX)
Alteración de los cromosomas sexuales con presencia de tres cromosomas X y un fenotipo femenino.
Alrededor de 1/1.000 mujeres aparentemente normales tienen un cariotipo 47,XXX, aunque rara vez presentan anomalías físicas. A veces sufren esterilidad o trastornos menstruales. Pueden tener una ligera alteración mental con puntuaciones medias del CI inmediatamente inferiores a 90 y, en comparación con sus hermanas, tienden a tener problemas escolares. El riesgo aumenta al hacerlo la edad de la madre y el cromosoma X adicional suele proceder de la madre.
Alteraciones raras del cromosoma X
Se han descrito mujeres con cariotipos 48,XXXX y 49,XXXXX, aunque su número es muy escaso. El fenotipo no es siempre constante. El riesgo de retraso mental y de malformaciones congénitas aumenta notablemente al hacerlo el número de cromosomas X, sobre todo cuando su número es>3. El desequilibrio genético durante las primeras fases de la vida embrionaria, antes de la inactivación de X, podría ser la causa de las alteraciones del desarrollo.
Síndrome de Klinefelter (47,XXY)
Alteración de los cromosomas sexuales con presencia de dos o más cromosomas X y uno Y, con fenotipo masculino.
El síndrome de Klinefelter afecta a 1 de cada 800 RN varones. En el 60% de los casos, el cromosoma X adicional es de procedencia materna.
Los pacientes tienden a ser altos, con brazos y piernas desproporcionadamente largos. A menudo tienen unos testículos pequeños y duros, y alrededor de la tercera parte desarrollan ginecomastia. Suelen alcanzar la pubertad a la edad habitual, pero a menudo su vello facial es escaso. Muestran predisposición a sufrir dificultades de aprendizaje y muchos tienen déficit del QI verbal, del procesamiento auditivo y de la lectura. Las variaciones clínicas son grandes y muchos varones 47,XXY tienen un aspecto e inteligencia normales, de forma que su alteración sólo se diagnostica durante un estudio por esterilidad (probablemente todos los varones 47,XXY son estériles) o en revisiones citogenéticas efectuadas en poblaciones normales. Se han hecho seguimientos de niños detectados en esta última forma sin que se haya observado una mayor incidencia de homosexualidad entre ellos. El desarrollo testicular varía entre túbulos hialinizados y no funcionantes a otros con cierta producción de espermatozoides; la excreción urinaria de hormona estimulante del folículo suele estar aumentada.
Variantes. En el 15% de los casos existe mosaicismo. Algunas personas afectadas tienen 3, 4 e incluso 5 cromosomas X, además del Y. En general, la gravedad del retraso mental y de las malformaciones aumenta a medida que lo hace el número de cromosomas X.
Síndrome 47,XYY
Alteración de los cromosomas sexuales con presencia de dos cromosomas Y y uno X, con fenotipo masculino.
El síndrome 47,XYY afecta a alrededor de 1 de cada 1.000 varones, cuya talla tiende a ser más alta que la media mientras que su CI muestra una disminución de 10 a 15 puntos en relación con los de su familia. Las alteraciones físicas son escasas, pero sí puede haber leves problemas de comportamiento, hiperactividad, trastornos por déficit de la atención y problemas de aprendizaje.
ESTADOS INTERSEXUALES
Situaciones en las que el aspecto de los genitales externo es ambiguo o distinto al del sexo cromosómico o gonadal de la persona.
(V. también Hiperplasia suprarrenal congénita, cap. 269.)
Etiología y clasificación
Los genitales se forman durante los primeros 3meses de la gestación gracias a una serie de acontecimientos iniciados por el cariotipo fetal y en los que intervienen en gran medida los esteroides sexuales. Las alteraciones de esta cascada pueden dar lugar a ambigüedades o disparidades genitales que se manifiestan por los estados intersexuales. La histología gonadal es la mejor base para su clasificación.
Seudohermafroditismo femenino. Tienen ovarios y genitales internos femeninos pero sus genitales externos son ambiguos; genéticamente son mujeres normales con un cariotipo 46,XX. La ambigüedad de los genitales externos se debe a la exposición a cantidades excesivas de andrógenos en la vida intrauterina. Los andrógenos responsables pueden ser exógenos (p. ej., progesterona administrada a la madre para evitar el aborto) pero lo más frecuente es que sean endógenos, debidos, por ejemplo, a un bloqueo enzimático en la esteroidogénesis por alteraciones genéticas del cromosoma6 (virilización suprarrenal o síndrome adrenogenital; v. también Virilización suprarrenal, cap. 9).
Seudohermafroditismo masculino. El tejido gonadal del paciente es únicamente testicular y, en general, su cariotipo es 46,XY. Sin embargo, los genitales externos suelen ser ambiguos aunque muy variables e incluso pueden ser totalmente femeninos, como sucede en la forma completa del síndrome de feminización testicular (síndrome de insensibilidad a los andrógenos). La etiología es compleja pero, en términos generales, el trastorno se debe a una producción inadecuada de andrógenos, a una respuesta inadecuada a estos o a una persistencia de elementos müllerianos, tal como se muestra en la tabla 261-5.

Hermafroditismo verdadero. Tienen tanto tejido testicular como ovárico y una mezcla de estructuras genitales masculinas y femeninas que depende del tejido gonadal predominante. En Estados Unidos, la mayoría de los hermafroditas verdaderos tienen un cariotipo 46,XX, pero el patrón puede ser muy variable. En raras ocasiones, los hermafroditas verdaderos tienen unos genitales externos totalmente masculinos.
Los pacientes con displasia gonadal mixta tienen tanto tejido testicular como tejido gonadal primitivo, denominados cintillas. El cariotipo de estos pacientes suele ser un mosaico 46,XY/45,XO y sus genitales son ambiguos. La talla de los adultos tiende a ser baja. Cuando las cintillas son bilaterales, el trastorno recibe el nombre de disgenesia gonadal pura y el fenotipo suele ser femenino.
Diagnóstico
En los pacientes con genitales ambiguos, tanto en los que tienen un fenotipo femenino y gónadas palpables como en los que son fenotípicamente masculinos pero con gónadas no palpables, ha de procederse a un estudio para valorar su intersexualidad. En general, en los varones con hipospadias de grado menor no suele ser necesaria esta valoración, sobre todo si los testículos están en las bolsas y su palpación es normal.
El diagnóstico de los RN afectados es urgente, no sólo para establecer su sexo (de especial importancia desde el punto de vista social), sino para identificar la pérdida de sal que puede acompañar a la virilización suprarrenal (v. también cap. 9) antes de que se desarrolle una hiponatremia potencialmente mortal. Debe procederse a la extracción inmediata de una muestra de sangre para determinación del cariotipo, aunque los resultados de ésta tardarán varios días. Entre tanto, la ecografía pélvica o una genitografía pueden demostrar la presencia de elementos müllerianos. Si se sospecha una virilización suprarrenal porque las gónadas no son palpables y se encuentran elementos müllerianos persistentes, deberá hacerse un control estrecho de los electrolitos para evitar la hiponatremia, midiendo la 17-hidroxiprogesterona sérica para documentar el bloqueo enzimático. En los casos intersexuales dudosos pueden ser necesarias la laparoscopia o una exploración quirúrgica con biopsia gonadal para establecer el diagnóstico definitivo. Si es posible, la asignación del sexo no debe retrasarse más allá de los primeros días de la vida.
Tratamiento
La asignación del sexo adecuado es de la máxima importancia. En general, a los seudohermafroditas femeninos se les asigna ese mismo sexo, mientras que en el caso de los seudohermafroditas masculinos la asignación depende de su desarrollo genital y de la actividad hormonal. En cuanto a los hermafroditas verdaderos, lo mejor es asignar el sexo más adecuado a su desarrollo genital, aunque la mayoría han sido reconstruidos como varones, lo que constituye una opción atractiva si el niño tiene testículos normalmente descendidos que pueden proporcionar una función hormonal al llegar a la pubertad. Los seudohermafroditas masculinos con un síndrome de feminización testicular totalmente desarrollado han de ser catalogados como mujeres, pero en otros muchos casos, lo mejor es asignar el sexo masculino. En los casos limítrofes, la administración de 1 o 2 ciclos de propionato de testosterona (en aceite) a dosis de 25 mg i.m. ayuda a determinar la capacidad de los genitales para responder a los andrógenos, lo que constituye un requisito esencial para catalogar al paciente como varón.
En el caso de la disgenesia gonadal mixta, lo mejor es asignar a los pacientes al sexo femenino, no sólo debido a su corta talla, sino también porque sus gónadas tienden a desarrollar tumores (gonadoblastoma). Se recomienda, por tanto, una reconstrucción precoz de los genitales externos con gonadectomía. Los pacientes con disgenesia gonadal pura tienen fenotipo femenino y deben ser criados como mujeres.
El momento óptimo para la reconstrucción de los genitales es variable. En las pacientes asignadas al sexo femenino que no tienen virilización suprarrenal se procede a la resección del clítoris de gran tamaño lo antes posible para facilitar su aceptación como mujeres por parte de la familia. En las que sufren virilización suprarrenal, se puede esperar unos meses hasta que se hayan estabilizado desde un punto de vista endocrinológico mediante un tratamiento con esteroides. En el caso de la reconstrucción vaginal, lo mejor es esperar hasta la pubertad, dada la elevada incidencia de estenosis que se produce cuando la intervención se hace en etapas más tempranas de la vida. La corrección del hipospadias en los varones suele hacerse cuando el niño tiene entre uno y dos años de edad (v. Pene y uretra, antes).