
300 / FARMACODINÁMICA
Estudio de los efectos bioquímicos y fisiológicos de los fármacos y de sus mecanismos de acción.
Muchos fármacos producen una respuesta farmacológica por medio de la interacción (unión) con macromoléculas específicas, por lo general proteínas complejas en las membranas celulares o en el interior de las células. Algunos fármacos interaccionan directamente con sustancias endógenas o exógenas de naturaleza no proteica, como ciertos antineoplásicos con ácidos nucleicos, fármacos quelantes con iones (p. ej., edetato cálcico disódico, dimercaprol, deferoxamina) o antiácidos neutralizantes químicos del ácido gástrico.
INTERACCIONES FÁRMACO-RECEPTOR
Muy pocos fármacos (o ninguno) muestran una especificidad absoluta, pero la mayoría actúan de manera selectiva; por ejemplo, la atropina inhibe los efectos de la acetilcolina sobre las glándulas exocrinas y la musculatura lisa, pero no sobre el músculo esquelético. Las acciones características de muchos de estos fármacos selectivos son consecuencia de su interacción fisicoquímica con componentes del organismo (es decir, lugares de reconocimiento de los fármacos), denominados convencionalmente receptores. Los receptores fisiológicos son macromoléculas responsables de las señales químicas inter e intracelulares. Las moléculas que se unen a los receptores se denominan ligandos. Cuando un ligando (hormona, neurotransmisor, mensajero intracelular o incluso un ligando exógeno: un fármaco) se une a un receptor, la función celular se modifica (v. tabla 300-1). Cada ligando puede unirse a múltiples subtipos de receptor. Los receptores activados directa o indirectamente pueden regular diferentes procesos bioquímicos celulares (p. ej., flujo de iones, fosforilación de proteínas, transcripción deADN). En muchos casos, los receptores localizados en la membrana celular están acoplados, a través de proteínas unidas a nucleótidos de guanina (proteínas G), a varios sistemas efectores que regulan moléculas que actúan como segundos mensajeros dentro de la célula.
Los receptores son dinámicos, es decir, están bajo la influencia tanto de factores externos como de mecanismos regulatorios intracelulares. La regulación al alza (up-regulation) y a la baja (down-regulation) de los receptores son fenómenos de gran importancia para entender ciertos cambios clínicamente relevantes de los tratamientos prolongados con fármacos (desensibilización, taquifilaxia, tolerancia, resistencia adquirida, hipersensibilidad tras la retirada).
Dentro de la macromolécula del receptor, existen regiones específicas a las que se unen los ligandos, que reciben el nombre de sitios de reconocimiento. Así, un fármaco podría interaccionar con el mismo sitio de reconocimiento que un agonista endógeno (hormona o neurotransmisor) o bien con un sitio distinto -pero adyacente- dentro del mismo receptor. Los agonistas que se unen en sitios diferentes se denominan agonistas alostéricos. También se cree que existe una fijación «inespecífica»: no todos los lugares moleculares a los que se fijan los fármacos son propiamente receptores (p. ej., proteínas plasmáticas).
La teoría del receptor, basada en la ley de acción de masas, puede compararse a los análisis cinéticos de la interacción e inhibición entre una enzima y un sustrato. En este marco de referencia se pueden estudiar y clasificar muchos mecanismos de acción de fármacos (p. ej., aspirina-inhibición de la prostaglandina-sintetasa, neostigmina-inhibición de la colinesterasa, deprenilo-inhibición de la monoaminooxidasa B). La teoría receptorial incluye los conceptos de afinidad (la probabilidad de que el fármaco esté ocupando el receptor en un momento determinado) y de actividad intrínseca (eficacia intrínseca), que expresa las complejas asociaciones entre las concentraciones de fármaco o ligando, el estado de activación o inhibición del receptor y las respuestas celulares o tisulares.
Las funciones fisiológicas (p. ej., contracción, secreción) están controladas por múltiples mecanismos regulados por receptores y, en consecuencia, pueden ser modulados por diferentes estímulos. Entre la interacción fármaco-receptor inicial y la respuesta final en el tejido u órgano tienen lugar varios pasos (acoplamiento con el receptor y numerosos sistemas de segundos mensajeros dentro de la célula). Además, la densidad de un tipo determinado de receptor y el grado de eficiencia (estímulo-respuesta) varía de un tejido a otro.
Las primeras teorías de la ocupación receptorial consideraban que una respuesta farmacológica era directamente proporcional al grado de ocupación receptorial; el efecto máximo se alcanza, según esa teoría, cuando todos los receptores están ocupados o inactivados. La moderna teoría alostérica y de proporción hace referencia a procesos cinéticos de la ocupación del complejo ligando-receptor (relación «en el receptor/fuera del receptor»), a los posibles estados de activación del receptor («activo/inactivo») y la falta de proporcionalidad aparente entre la ocupación del receptor-ligando y la respuesta del tejido o del órgano. Los modelos teóricos actuales consideran variaciones en la eficacia de la transducción de señales (mecanismos intracelulares de amplificación de la señal), hacen referencia al «ahorro» de receptores e incluyen la existencia de agonistas parciales y agonistas inversos (v. más adelante).
Los fármacos agonistas interaccionan con el receptor alterando la proporción de receptores activados, modificando la actividad celular. Los agonistas convencionales aumentan la proporción de receptores activados, los agonistas inversos la reducen. Numerosas hormonas y neurotransmisores (acetilcolina, histamina o noradrenalina) y varios fármacos (morfina, fenilefrina e isoproterenol) acúan como agonistas.
Muchos otros tipos de fármacos interaccionan de manera selectiva con receptores, pero no inician la secuencia de acontecimientos que conducen al efecto observado. Reducen, además, las acciones de otras sustancias (agonistas) que actúan sobre el receptor. Estos fármacos se denominan antagonistas: poseen afinidad, pero carecen de eficacia intrínseca.
Este dualismo agonista-antagonista se complica más por el hecho de que algunos análogos estructurales de las moléculas agonistas muestran frecuentemente una mezcla de propiedades agonistas y antagonistas; estos fármacos se denominan agonistas parciales o agonistas de baja eficacia. Por ejemplo, el isoproterenol es un potente agonista de los receptores b-adrenérgicos, mientras que el prenalterol es un agonista parcial de esos receptores en algunos tejidos. Un fármaco puede actuar como agonista parcial en un tejido y como agonista completo en otro.
Los antagonistas se clasifican como reversibles o irreversibles, dependiendo de su interacción con los receptores. Los antagonistas reversibles se disocian fácilmente de su receptor; los antagonistas irreversibles forman un enlace químico estable con el receptor (p. ej., alquilación). Otros se disocian lentamente del receptor (antagonistas seudoirreversibles). Si el antagonista se fija en el mismo sitio del receptor que el agonista (la unión es mutuamente excluyente), se trata de un antagonismo competitivo. Los antagonistas no competitivos pueden fijarse simultáneamente, pero la unión del antagonista reduce o previene la acción del agonista. En el caso de antagonismo competitivo reversible, el agonista y el antagonista forman combinaciones de corta duración con el receptor, alcanzando un equilibrio entre las moléculas de agonista, de antagonista y de receptor. Este tipo de antagonismo puede ser remontado aumentando las concentraciones del agonista. Por ejemplo, la naloxona -un antagonista de receptores opioides estructuralmente similar a la morfina, con escasa o nula actividad opiácea- bloquea los efectos de la morfina cuando se administra antes o después de ella. Sin embargo, el antagonismo competitivo de la naloxona puede ser superado administrando más morfina.
RELACIONES DOSIS-RESPUESTA
Correspondencia entre la cantidad de fármaco administrada y la magnitud de la respuesta evocada.
El conocimiento de las relaciones dosis-respuesta es fundamental en la toma de decisiones terapéuticas y en la farmacología experimental. Los datos dosis-respuesta se representan gráficamente en dos dimensiones, expresando el efecto medido (respuesta) en las ordenadas (variable dependiente) y la dosis o una función de la dosis (p. ej., su log) en el eje de abscisas (variable independiente). Puesto que el efecto farmacológico está en función de la dosis (o concentración) y del tiempo, esta representación proporciona una relación dosis-respuesta independiente del tiempo. Habitualmente, los efectos se presentan como máximos, registrados en el momento del pico de efectos, o en estado de equilibrio estacionario (durante perfusiones i.v. continuas). Las acciones farmacológicas pueden cuantificarse a distintos niveles: molecular, celular, tisular, orgánico, de un sistema de órganos o de todo el organismo.
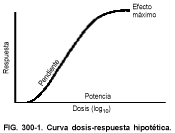
La figura 300-1 ilustra las cuatro características variables de una curva dosis-respuesta hipotética: 1) la potencia (emplazamiento de la curva en el eje de la dosis), 2) la eficacia máxima o techo del efecto (mayor respuesta posible del sistema que se está midiendo), 3) la pendiente (cambio de la respuesta por unidad de dosis) y 4) la variación biológica (variación en la magnitud de la respuesta entre los distintos sujetos de la misma población a los que se ha administrado la misma dosis). El estudio de la representación gráfica de las curvas dosis-respuesta de fármacos estudiados bajo las mismas condiciones experimentales resulta muy útil para comparar los perfiles farmacológicos de dichos fármacos (v. fig. 300-2).
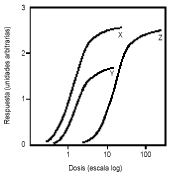
FIG. 300-2. Comparación de las curvas dosis-respuesta para los fármacos X, Y y Z. El fármaco X, que tiene la mayor actividad biológica por dosis equivalente, es más potente que los fármacos Y y Z. Los fármacos X y Z tienen igual eficacia, como indica el hecho de que poseen el mismo efecto máximo (techo). El fármaco Y es más potente que el Z, pero su efecto máximo es menor.